ABSTRACT
Background
Cancer is the world’s leading cause of death; more than ten million people die from cancer each year. Rate of morbidity and mortality is increasing day by day. As a result, utilizing chemoinformatics techniques, a novel series of Methylenedioxyphenyl linked with 3,4,5- trimethoxyphenyl/2,5- dimethoxyphenyl and pyrimidine has been designed, docked and in-silico predictions of pharmacokinetic, and toxicological parameters.
Materials and Methods
A novel class of Methylenedioxyphenyl derivatives was docked by using AutoDock Vina software to reveal the interaction of these derivatives with the active site topoisomerase-II with PBD ID 5GWK. In addition to the above, the ADME studies were performed using Swiss ADME software and the toxicity parameters using the pkCSM online tool.
Results
Many derivatives were found to be more active than the standard drug etoposide in docking studies. Binding interaction pattern of selected compounds were studied with the active sites of DNA topoisomerase-II (PBD ID 5GWK) by docking simulation. The results of the screening revealed that compounds B9, B12, B13, B15, B16, B17, B18, B22, and B23 were more active candidates of the series.
Conclusion
Molecular docking and ADME/Tox properties of new methylenedioxyphenyl derivatives have been described. As a result, nine compounds from the intended series showed improved docking scores with suitable ADME/Tox properties and satisfactory topo II inhibiting activity, thus these compounds may be the effective inhibitors of topoisomerase IIα enzyme.
INTRODUCTION
Topoisomerases are widely distributed enzymes that play important roles in biological processes such replication of DNA, transcription, recombination, and the repair of DNA that has been supercoiled, relaxed, catenated, or knotted. Topoisomerases of all types demonstrate their biological activities by catalyzing DNA breakage and relegation. Antibacterial and anticancer medications have been shown to target.1,2 DNA topoisomerases therapeutically. The precise binding locations and mechanisms of topoisomerase poisons have been identified by structural studies of DNA-topoisomerase and inhibitor ternary complexes. Human Topoisomerase (topo) II are available in two isoforms i.e. topoisomerase IIα and topoisomerase IIβ. According to the tissue type and replicative activity, both of them carry out similar tasks at varying levels.3,4 Human topoisomerase IIα and topoisomerase IIβ are dispersed throughout different cells and tissues, although they share a similar tertiary structure and basic sequence. They also reveal a variety of cellular roles; topoisomerase II is mainly found in the nuclear plasma and is over expressed in cells that are actively dividing. Topo II appears to be involved in transcriptional control, cell development, and cell differentiation, but it is not necessary for cell survival and proliferation. Topoisomerase IIβ does not relax negatively supercoiled plasmids despite the fact that topoisomerase IIα more slowly than the positive supercoiled plasmid. All these findings suggest that topoisomerase IIα is the most attractive target for novel anticancer medications. Therefore, screened topoisomerase IIα inhibitors have drawn a lot of attention in cancer therapy because they may offer a more focused method of treating the extremely proliferative.5–7
In recent times, the best forms of binding interactions of compounds and target proteins have been determined using the computational protein ligand docking. The depiction of an ideal combination that might be anticipated from the structure of a target protein and the prospective therapeutic molecule is made possible by the ligand docking approach. The main focus of the structure based virtual screening process on the therapeutic targets three dimensional information for the docking study. Docking techniques are utilized to choose the hits that show chemical, structural, and electrical features. In silico method or experimental data helps to learn more about the detail of the target protein. All of these benefits have promoted the use of computational approaches in drug discovery to investigate lead compounds for target proteins.8,9
The homodimer structure of human topoisomerase IIα is 1531 amino acids long, with four parts N-gate, C-gate, DNA-gate and C-Terminal Domains-making up its monomer (CTD).10 This enzyme’s X-ray crystal structure can be obtained from the Protein Database with PDB: 5GWK (www.rcsb.org) and we conducted in-silico research using this structure.11 In this work, we propose new anti-cancer compounds that have the necessary qualities to be potential oral human topo II inhibitors by predicting the binding modes and calculating the ADME/Tox characteristics.
MATERIALS AND METHODS
Selection of PDB structure of DNA topoisomerase II
The protein data bank contains many structures of topo II in complex with small molecules, agonists/antagonists etc. The structure of topo IIα complexed with DNA and etoposide (PDB ID: 5GWK) was employed for present study. The PDB structure was chosen because of its high resolution and complexed ligand. The Table 1 shows the details of selected PDB structure.
| Name of protein | Human topoisomerase II alpha in complex with DNA and etoposide |
|---|---|
| PDB ID | 5GWK |
| Resolution | 3.15 Å |
| Complexed ligand | 5S,5aR,8aR,9R)-9-(4-hydroxy- 3,5-dimethoxyphenyl)-8-oxo- 5,5a,6,8,8a,9-hexahyd rofuro[3’,4’:6,7]naphtho[2,3-d][1,3]- dioxol-5-yl 4,6-O-[(1R)- ethylidene]-beta-D- glucopyranoside |
| R-value free | 0.244 |
| R-value work | 0.203 |
| Method | X-ray diffraction |
Molecular docking analysis
Molecular docking of designed molecules into the active position of topo IIα complexed with DNA and etoposide were carried out using AutoDock Tool (ADT) software.12 In the preparation process, polar hydrogen was added, the partial charges calculation was corrected and Gasteiger charges for each atom were determined. The three-dimensional structure of protein 5GWK is displayed in Figure 1. All compounds were drawn by ChemDraw Ultra software (Figure 2) and energy was minimized by MM2 tool of Chem3D Ultra. They are used as ligand. AutoDock tools was used to calculate the torsion of ligands.13,14 The grid size was defined on the bases of essential amino acid residues for binding pocket (reference). Using the information about protein, ligand and grid box parameters docking studies were conducted. Docking results from this work would be useful in understanding the inhibitory mode along with quickly and precisely forecasting the actions of new therapeutic agents based on docking scores. This research provided the vital information about the orientation of the inhibitor in the binding pocket of the target protein, given the significance of intercalation binding method for this experiment. The docking poses were analyzed and visualized using auto dock vina and discovery studio.15,16
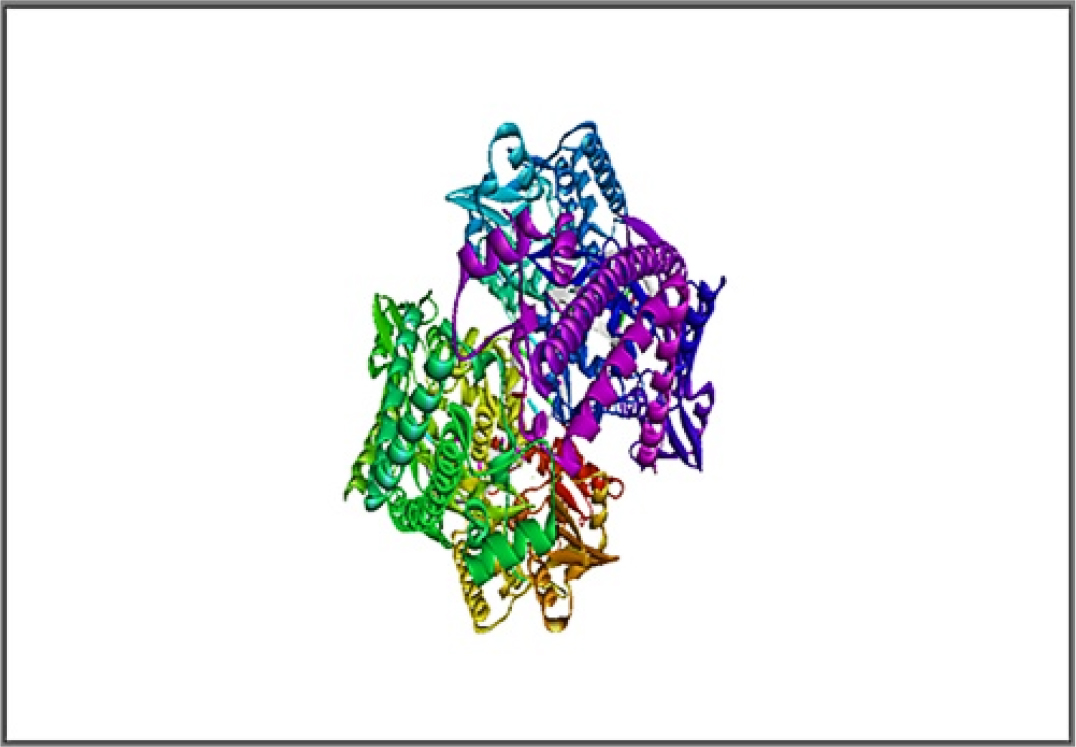
Figure 1:
Topoisomerase IIα complexed a three-dimensions with etoposide and DNA (PDB ID-5GWK).
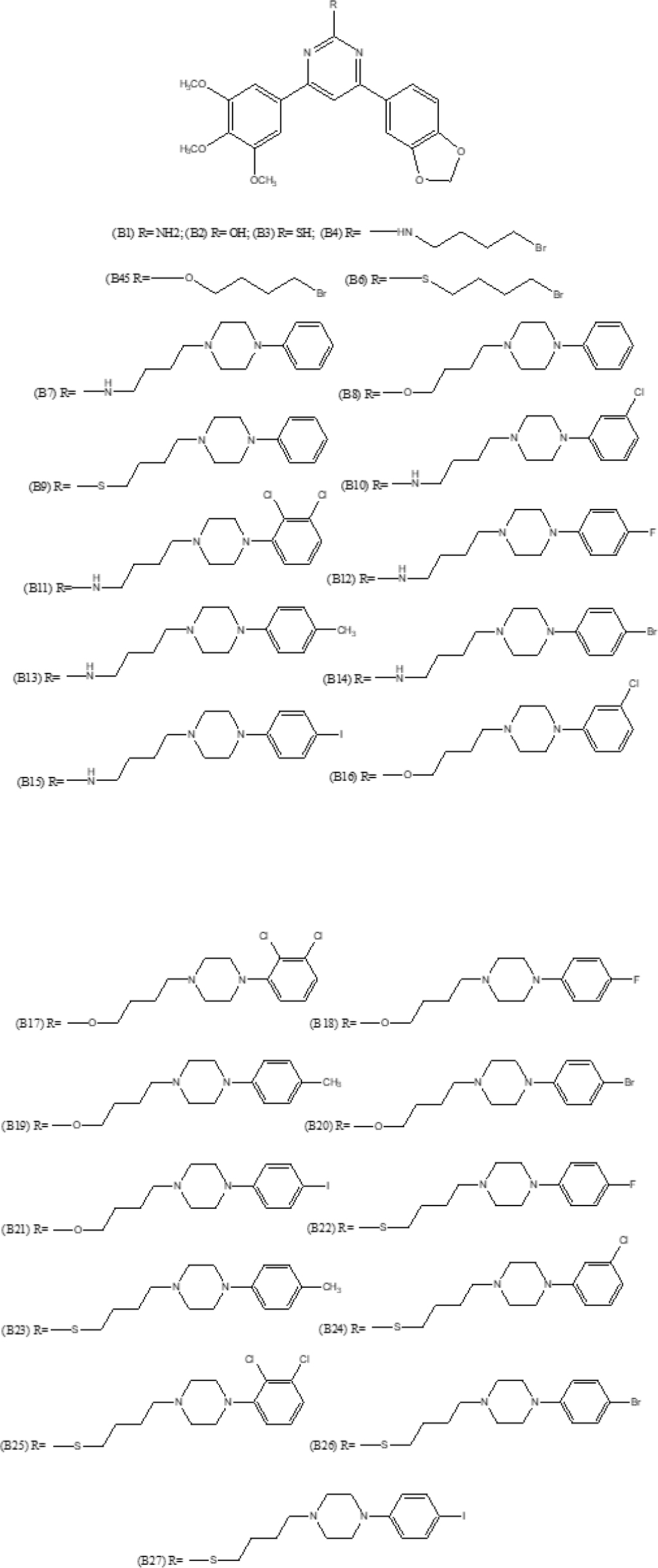
Figure 2:
Structures of pyrimidine containing methylenedioxyphenyl derivatives.
ADMET Analysis
Steps involved
- Open SwissADME tool.
- Molecular Sketcher – Draw, edit, import, open molecular structure from files.
- Incorporate the sketched structure into the SMILES file. The double arrow button allows you to import data from the databases, create a native file, or design the two dimensional structure that will be added to the list.
- One molecule per line in the SMILES file. Molecules can be pasted or written directly into the SMILES format, or they can be entered using the molecular sketcher.
- When you’re ready to submit the list of molecules, click the “Run” button to begin the computations.
- The result/output of calculation is loaded in the same Web page.
- The calculated results are downloaded as MS-excel sheet.
Toxicity Analysis
In order to calculate the pharmacokinetic and physicochemical characteristics during the process of drug discovery, the standardized pharmacokinetic was established.22 Chemdraw is used to create the structure of the compounds, which is then imported into SMILES through the website’s user interface (http://swissadme.ch/). In the pkCSM online tool, these compounds SMILES were loaded.23 Toxicological predictions were performed using the pkCSM programme. The AMES toxicity, maximum tolerated dose for humans, oral acute toxicity, oral chronic toxicity and minnow toxicity were all chosen using the toxicity mode of pkCSM online programme.
RESULTS
Results of docking studies
The molecular docking study was done on the ligand-protein interactions and binding pattern of ligands into the active sites pocket of the Topo IIα enzyme. The designed ligands were docked into the binding pocket of topoisomerase II alpha. The ligand was kept flexible and the receptor was made rigid throughout docking studies. This analysis highlights the significance of the residues in the binding sites that were reported to bind with most of the ligands. The compounds were ranked based on their binding affinity. More negative binding affinity indicates the stronger binding. The hydrogen bond and non-bonded interactions were studied for all of ligands. Table 2 provides the binding affinities of all docked ligands, whereas Table 3 displays the interactions involving hydrogen and hydrophobic bonds. The amino acid binding interactions of better docking score ligands and co-crystal ligand were shown in Figure 3. The co-crystallized ligand achieved binding affinity of -10.2 and established hydrogen bond interactions with ASP463 and MET766. It exhibited hydrophobic interactions with ARG487, MET762, PRO803, GLY462, DG13, DC8, and DT9.
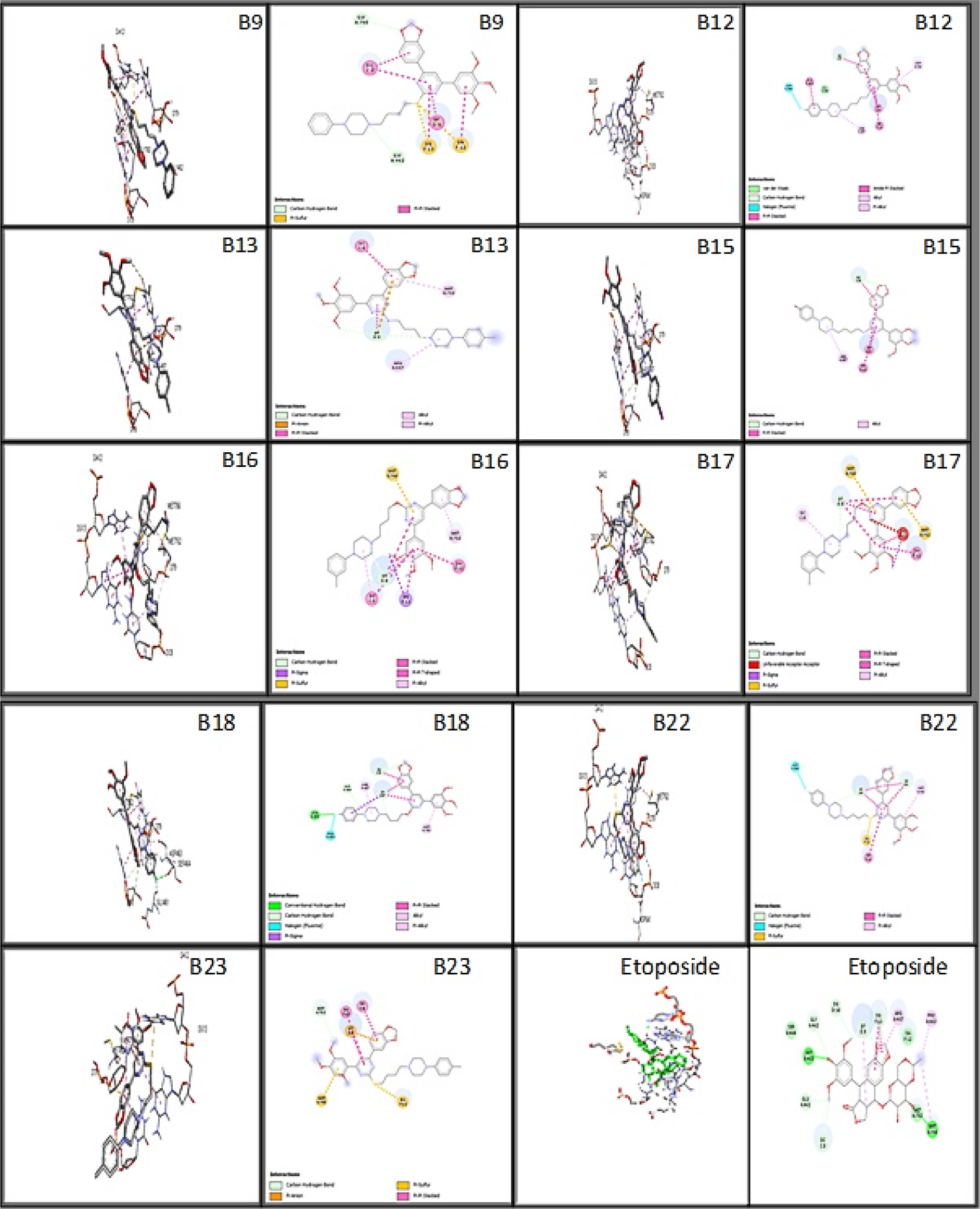
Figure 3:
The best designed ligands’ 3D and 2D binding interactions with the co-crystallized ligand (Etoposide) on the 5GWK enzyme target.
| Sl. No. | Molecule Name | Binding affinity (kcal/mol) |
|---|---|---|
| 1 | B1 | -9.9 |
| 2 | B2 | -9.9 |
| 3 | B3 | -9.9 |
| 4 | B4 | -9.6 |
| 5 | B5 | -9.1 |
| 6 | B6 | -9.1 |
| 7 | B7 | -9.4 |
| 8 | B8 | -9.9 |
| 9 | B9 | -10.4 |
| 10 | B10 | -9.9 |
| 11 | B11 | -9.7 |
| 12 | B12 | -11.0 |
| 13 | B13 | -10.6 |
| 14 | B14 | -10.0 |
| 15 | B15 | -10.8 |
| 16 | B16 | -10.4 |
| 17 | B17 | -10.7 |
| 18 | B18 | -11.3 |
| 19 | B19 | -9.9 |
| 20 | B20 | -9.8 |
| 21 | B21 | -9.9 |
| 22 | B22 | -10.9 |
| 23 | B23 | -10.3 |
| 24 | B24 | -10.2 |
| 25 | B25 | -9.9 |
| 26 | B26 | -9.7 |
| 27 | B27 | -9.6 |
| 28 | co-crystallized ligand (Etoposide) | -10.2 |
| Sl. No. | Molecule Name | Hydrogen and Hydrophobic bond interactions |
|---|---|---|
| 01 | B9 | GLY462, GLY760, DC8, DT9, DA12, DG13 |
| 02 | B12 | GLU461, GLY462, ARG487, ASP541, MET762, DC8, DT9, DG13 |
| 03 | B13 | ARG487, MET762, DC8, DT9 |
| 04 | B15 | ARG487, DC8, DT9, DG13 |
| 05 | B16 | MET762, MET766, DC8, DT9, DA12, DG13 |
| 06 | B17 | MET762, MET766, DC8, DT9, DA12, DG13 |
| 07 | B18 | GLU461, ASP463, SER464, ARG487, MET762, DC8, DT9 |
| 08 | B22 | ASP541, MET762, DC8, DT9, DA12, DG13 |
| 09 | B23 | MET762, MET766, DC8, DT9, DA12, DG13 |
| 10 | Co-crystallized ligand (Etoposide) | ASP463, MET766, ARG487, MET762, PRO803, GLY462, DG13, DC8, DT9 |
Results of ADME studies
ADME assays are performed to evaluate the druggability of suggested compounds before synthesis. The SwissADME program helps to predict pharmacokinetic and druglikeness characteristics of the proposed compound, and the results are displayed in Tables 4, 5, and 6. All compounds, with the exception of compounds B1, B2, and B3, have molecular weights larger than 500 g/mol, according to the physio-chemical properties. 28 heavy atoms are found in compounds B1, B2, and B3, 33 heavy atoms are found in compounds B4, B5, and B6, 44 heavy atoms are found in compounds B7, B8, and B9, and 46 heavy atoms are found in compounds B11, B17, and B25. The remaining compounds all include 45 heavy atoms. All compounds have 24 aromatic heavy atoms, except B1, B2, B3, B4, B5, and B6 have 18 aromatic heavy atoms. The majority of compounds having 12 rotatable bonds but compounds B1, B2, B3 have 5 and compound B4, B5, B6 have 10 rotatable bonds. The H-bond donor groups are present in compounds B1-2, B4, B7, and B10-15. Compounds B1, B3, B4, and B6 each contain seven H-bond acceptors, while compounds B2, B5, B7, B9-11, B13-14, and B25-27 each have eight. Compounds B8, B12, B16-17, B19-22 each have nine, and compound B18 has ten.
| Comp ound | Formula | MW | Heavy atoms | Aromatic heavy atoms | Fraction Csp3 | Rotatable bonds | H-bond acceptors | H-bond donors | MR | TPSA |
|---|---|---|---|---|---|---|---|---|---|---|
| B1 | C20H19N3O5 | 381.38 | 28 | 18 | 0.2 | 5 | 7 | 1 | 102.85 | 97.95 |
| B2 | C20H18N2O6 | 382.37 | 28 | 18 | 0.2 | 5 | 8 | 1 | 100.47 | 92.16 |
| B3 | C20H18N2O5S | 398.43 | 28 | 18 | 0.2 | 5 | 7 | 0 | 105.69 | 110.73 |
| B4 | C24H26BrN3O5 | 516.38 | 33 | 18 | 0.33 | 10 | 7 | 1 | 130.04 | 83.96 |
| B5 | C24H25BrN2O6 | 517.37 | 33 | 18 | 0.33 | 10 | 8 | 0 | 127.23 | 81.16 |
| B6 | C24H25BrN2O5S | 533.43 | 33 | 18 | 0.33 | 10 | 7 | 0 | 132.45 | 97.23 |
| B7 | C34H39N5O5 | 597.7 | 44 | 24 | 0.35 | 12 | 8 | 1 | 177.95 | 90.44 |
| B8 | C34H38N4O6 | 598.69 | 44 | 24 | 0.35 | 12 | 9 | 0 | 175.14 | 87.64 |
| B9 | C34H38N4O5S | 614.75 | 44 | 24 | 0.35 | 12 | 8 | 0 | 180.37 | 103.71 |
| B10 | C34H38ClN5O5 | 632.15 | 45 | 24 | 0.35 | 12 | 8 | 1 | 182.96 | 90.44 |
| B11 | C34H37Cl2N5O5 | 666.59 | 46 | 24 | 0.35 | 12 | 8 | 1 | 187.97 | 90.44 |
| B12 | C34H38FN5O5 | 615.69 | 45 | 24 | 0.35 | 12 | 9 | 1 | 177.91 | 90.44 |
| B13 | C35H41N5O5 | 611.73 | 45 | 24 | 0.37 | 12 | 8 | 1 | 182.92 | 90.44 |
| B14 | C34H38BrN5O5 | 676.6 | 45 | 24 | 0.35 | 12 | 8 | 1 | 185.65 | 90.44 |
| B15 | C34H38IN5O5 | 723.6 | 45 | 24 | 0.35 | 12 | 8 | 1 | 190.67 | 90.44 |
| B16 | C34H37ClN4O6 | 633.13 | 45 | 24 | 0.35 | 12 | 9 | 0 | 180.15 | 87.64 |
| B17 | C34H36Cl2N4O6 | 667.58 | 46 | 24 | 0.35 | 12 | 9 | 0 | 185.16 | 87.64 |
| B18 | C34H37FN4O6 | 616.68 | 45 | 24 | 0.35 | 12 | 10 | 0 | 175.09 | 87.64 |
| B19 | C35H40N4O6 | 612.72 | 45 | 24 | 0.37 | 12 | 9 | 0 | 180.1 | 87.64 |
| B20 | C34H37BrN4O6 | 677.58 | 45 | 24 | 0.35 | 12 | 9 | 0 | 182.84 | 87.64 |
| B21 | C34H37IN4O6 | 724.59 | 45 | 24 | 0.35 | 12 | 9 | 0 | 187.85 | 87.64 |
| B22 | C34H37FN4O5S | 632.74 | 45 | 24 | 0.35 | 12 | 9 | 0 | 180.32 | 103.71 |
| B23 | C35H40N4O5S | 628.78 | 45 | 24 | 0.37 | 12 | 8 | 0 | 185.33 | 103.71 |
| B24 | C34H37ClN4O5S | 649.2 | 45 | 24 | 0.35 | 12 | 8 | 0 | 185.38 | 103.71 |
| B25 | C34H36Cl2N4O5S | 683.64 | 46 | 24 | 0.35 | 12 | 8 | 0 | 190.39 | 103.71 |
| B26 | C34H37BrN4O5S | 693.65 | 45 | 24 | 0.35 | 12 | 8 | 0 | 188.07 | 103.71 |
| Compound | GI absorption | BBB permeant | Pgp substrate | CYP1A2 inhibitor | CYP2C19 inhibitor | CYP2C9 inhibitor | CYP2D6 inhibitor | CYP3A4 inhibitor | log Kp (cm/s) |
|---|---|---|---|---|---|---|---|---|---|
| B1 | High | No | Yes | Yes | No | Yes | Yes | Yes | -6.56 |
| B2 | High | No | No | Yes | No | Yes | Yes | Yes | -6.34 |
| B3 | High | No | No | Yes | Yes | Yes | Yes | Yes | -6.08 |
| B4 | High | No | Yes | No | Yes | Yes | Yes | Yes | -5.86 |
| B5 | High | No | Yes | No | Yes | Yes | Yes | Yes | -5.88 |
| B6 | High | No | Yes | No | Yes | Yes | Yes | Yes | -5.60 |
| B7 | High | No | Yes | No | Yes | No | No | No | -5.74 |
| B8 | High | No | Yes | No | Yes | Yes | Yes | No | -5.76 |
| B9 | High | No | Yes | No | Yes | Yes | No | No | -5.48 |
| B10 | High | No | Yes | No | Yes | No | No | No | -5.51 |
| B11 | High | No | Yes | No | Yes | No | Yes | No | -5.28 |
| B12 | High | No | Yes | No | Yes | No | Yes | No | -5.78 |
| B13 | High | No | Yes | No | Yes | No | No | No | -5.57 |
| B14 | High | No | Yes | No | Yes | No | No | No | -5.73 |
| B15 | High | No | Yes | No | Yes | No | No | No | -6.05 |
| B16 | High | No | Yes | No | Yes | Yes | Yes | No | -5.53 |
| B17 | High | No | Yes | No | Yes | No | Yes | No | -5.29 |
| B18 | High | No | Yes | No | Yes | Yes | Yes | No | -5.80 |
| B19 | High | No | Yes | No | Yes | Yes | No | No | -5.59 |
| B20 | High | No | Yes | No | Yes | No | Yes | No | -5.75 |
| B21 | High | No | Yes | No | Yes | No | Yes | No | -6.07 |
| B22 | Low | No | Yes | No | Yes | No | No | No | -5.52 |
| B23 | Low | No | Yes | No | Yes | No | No | No | -5.30 |
| B24 | Low | No | Yes | No | Yes | No | No | No | -5.24 |
| B25 | Low | No | Yes | No | Yes | No | No | No | -5.00 |
| B26 | Low | No | Yes | No | Yes | No | No | No | -5.47 |
| B27 | Low | No | Yes | No | Yes | No | No | No | -5.78 |
| Compound | Lipinski violations | Ghose | Veber | Egan | Muegge | Bioavailability Score |
|---|---|---|---|---|---|---|
| B1 | 0 | 0 | 0 | 0 | 0 | 0.55 |
| B2 | 0 | 0 | 0 | 0 | 0 | 0.55 |
| B3 | 0 | 0 | 0 | 0 | 0 | 0.55 |
| B4 | 1 | 2 | 0 | 0 | 1 | 0.55 |
| B5 | 1 | 1 | 0 | 0 | 1 | 0.55 |
| B6 | 1 | 3 | 0 | 0 | 1 | 0.55 |
| B7 | 1 | 3 | 1 | 0 | 1 | 0.55 |
| B8 | 1 | 3 | 1 | 0 | 1 | 0.55 |
| B9 | 1 | 3 | 1 | 0 | 2 | 0.55 |
| B10 | 1 | 3 | 1 | 0 | 2 | 0.55 |
| B11 | 1 | 4 | 1 | 1 | 2 | 0.55 |
| B12 | 1 | 3 | 1 | 0 | 2 | 0.55 |
| B13 | 1 | 3 | 1 | 0 | 2 | 0.55 |
| B14 | 1 | 3 | 1 | 0 | 2 | 0.55 |
| B15 | 1 | 3 | 1 | 0 | 2 | 0.55 |
| B16 | 1 | 3 | 1 | 0 | 2 | 0.55 |
| B17 | 1 | 4 | 1 | 1 | 2 | 0.55 |
| B18 | 1 | 3 | 1 | 0 | 2 | 0.55 |
| B19 | 1 | 3 | 1 | 0 | 2 | 0.55 |
| B20 | 1 | 3 | 1 | 0 | 2 | 0.55 |
| B21 | 1 | 3 | 1 | 0 | 2 | 0.55 |
| B22 | 1 | 4 | 1 | 1 | 2 | 0.55 |
| B23 | 1 | 4 | 1 | 0 | 2 | 0.55 |
| B24 | 1 | 4 | 1 | 1 | 2 | 0.55 |
| B25 | 1 | 4 | 1 | 1 | 2 | 0.55 |
| B26 | 1 | 4 | 1 | 1 | 2 | 0.55 |
| B27 | 1 | 4 | 1 | 1 | 2 | 0.55 |
As per the pharmacokinetic parameters, all compounds exhibited high Gastrointestinal (GI) absorption except B22 to B27 having low GI absorption. Compounds do not have BBB permeability. The majority of compounds showed no inhibition to Cytochrome P450 isomers CYP1A2 except B1, B2, and B3. There was no inhibition of the isomers of CYP2D6 by compounds B7, B9, B10, B13-15, B19, and B22-27. The rest of the compounds did not inhibit CYP3A4 except for compound B1, B2, B3, B4, B5, and B6. All compounds, with the exception of B1 and B2, were found to inhibit the cytochrome P450 isomer CYP2C19.Isomer CYP2C9 was inhibited by compounds B1-6, B8-9, B16, B18-19 and all other compounds showed no inhibition. Compounds B2, and B3 have no P-glycoprotein substrate.
In accordance with the prescribed Lipinski, Ghose, Veber criteria and bioavailability score, drug-likeness predictions were also made. The Lipinski filter is the original application of the “rule of five,” which asserts that a molecule is more likely to absorb or permeate if its mol. wt. (molecular weight) is less than 500 gm/mol, log P significant value is less than 5, and it has no more than 5 hydrogen bond donor and 10 hydrogen bond acceptor atoms. All Compounds have 1 Lipinski violation except B1, B2, and B3.
According to Ghose filter, the requirements for drug likeness are as follows: The reported mol. wt. ranging from 160-480, molar refractivity ranging from 40-130, and calculated logP values ranging from -0.4 to 5.6. The total atom count ranging from 20-70. Compounds were rejected with one, two, three or four violations in screening with Ghose rules except compound B1, B2 and B3. The designed compounds satisfied the requirements for evaluating drug likeness using Veber guidelines. Drug likeness constraints are described by Veber (GSK) regulation as Rotatable bond count 10 and PSA 140.
Muegge and colleagues developed an easy pharmacophore point filter that divides chemical matter into molecules that resemble drugs and non-drug-like substances. It is based on the fact that non drugs are often under functionalized. As a result, the filter requires a minimum quantity of well-characterized pharmacophore points in order to function. The synthesised compounds’ Muegge (Bayer) filter drug likeness assessment revealed that none of the compounds matched the criteria to pass the filter. Interestingly, the Muegge (Bayer) filter rejected all the compounds with one violation, with the exception of B1, B2, and B3. In Table 6, each chemical has the same bioavailability rating of 0.55.
After all the calculations were completed, a graphical output is represented in the form of Boiled Egg (as shown in Figure 4). The compounds in the BOILED-Egg can be intuitively evaluated for passive gastrointestinal absorption and brain penetration using the WLOGP-versus-TPSA referential position. The white part is for high possibility passive absorption by the gastrointestinal tract, whereas the yellow part is for high possibility brain penetration.
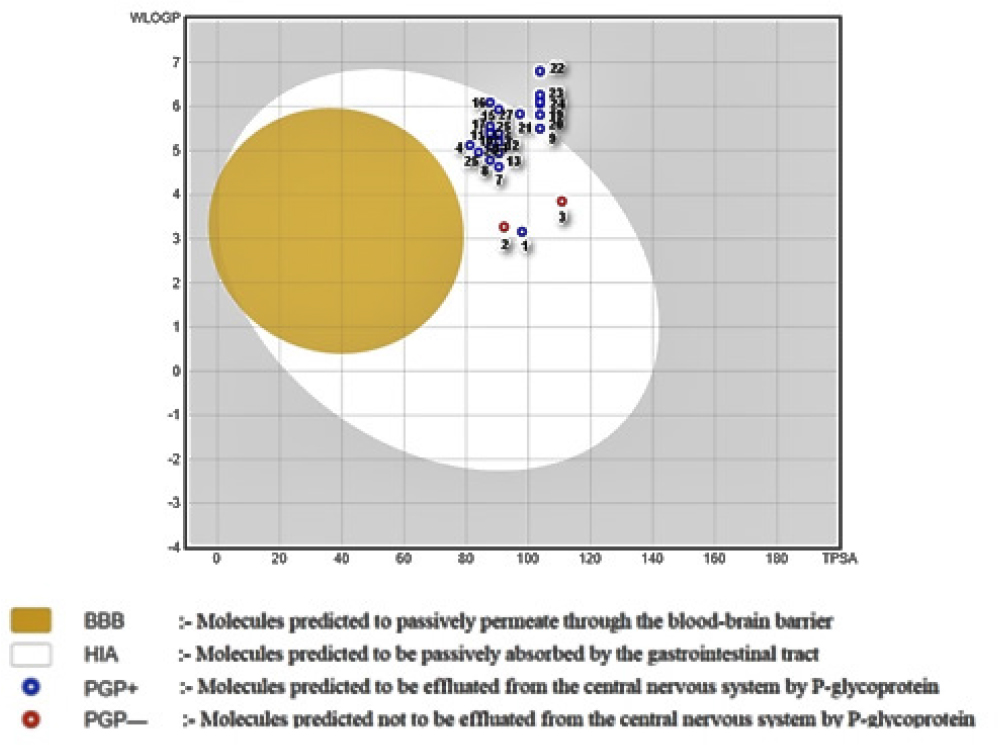
Figure 4:
A straightforward, graphical classification model for gastrointestinal and brain absorption for designed series.
Results of toxicity studies
A toxicity assessment is a method for determining the toxicity of molecules that are harmful to people, animals, or plants. In silico toxicity studies’ major objectives are to complement existing toxicological tests, direct toxicity testing, prioritise substances, and decrease the chance of late-stage failures in medication creation. In order to predict the toxicity of synthesized compound, the pkCSM online tool was used, in which various factors like hepatotoxicity, skin sensitization, AMES toxicity, minnow toxicity, carcinogenicity, and skin sensitization were predicted. All the predicted results are represented in Table 7.
| AMES toxicity | Max tolerated dose | hERG I Inhibitor | hERG II Inhibitor | Oral Rat Acute Toxicity (LD50) | Oral Rat Chronic Toxicity (LOAEL) | Hepato-toxicity | Skin Sensitization | T. Pyriformis Toxicity | Minnow Toxicity | |
|---|---|---|---|---|---|---|---|---|---|---|
| B1 | No | 0.203 | No | Yes | 2.721 | 0.995 | Yes | No | 0.289 | -0.026 |
| B2 | No | 0.476 | No | Yes | 2.78 | 1.157 | Yes | No | 0.286 | 0.038 |
| B3 | No | 0.272 | No | Yes | 2.991 | 0.901 | Yes | No | 0.296 | -0.397 |
| B4 | No | 0.911 | No | Yes | 2.379 | 0.483 | Yes | No | 0.286 | 0.504 |
| B5 | No | 0.904 | No | Yes | 2.175 | 0.627 | Yes | No | 0.285 | -1.637 |
| B6 | No | 0.908 | No | Yes | 2.3 | 0.439 | No | No | 0.285 | -1.441 |
| B7 | No | 0.461 | No | Yes | 2.816 | 1.581 | Yes | No | 0.285 | -0.739 |
| B8 | No | 0.774 | No | Yes | 2.95 | 0.374 | Yes | No | 0.285 | -1.883 |
| B9 | No | 0.765 | No | Yes | 2.86 | 0.161 | Yes | No | 0.285 | -2.213 |
| B10 | No | 0.461 | No | Yes | 2.763 | 1.511 | Yes | No | 0.285 | -0.84 |
| B11 | No | 0.477 | No | Yes | 2.728 | 1.439 | Yes | No | 0.285 | -1.155 |
| B12 | No | 0.453 | No | Yes | 2.735 | 1.494 | No | No | 0.285 | -0.599 |
| B13 | No | 0.445 | No | Yes | 2.795 | 1.559 | Yes | No | 0.285 | -0.748 |
| B14 | No | 0.462 | No | Yes | 2.76 | 1.509 | Yes | No | 0.285 | -1.111 |
| B15 | No | 0.459 | No | Yes | 2.764 | 1.52 | Yes | No | 0.285 | -1.251 |
| B16 | No | 0.775 | No | Yes | 2.919 | 0.114 | Yes | No | 0.285 | -2.05 |
| B17 | No | 0.784 | No | Yes | 2.904 | -0.088 | Yes | No | 0.285 | -2.365 |
| B18 | No | 0.757 | No | Yes | 2.903 | 0.044 | Yes | No | 0.285 | -1.711 |
| B19 | No | 0.763 | No | Yes | 2.927 | 0.331 | Yes | No | 0.285 | -1.958 |
| B20 | No | 0.775 | No | Yes | 2.991 | 0.093 | Yes | No | 0.285 | -2.321 |
| B21 | No | 0.773 | No | Yes | 2.919 | 0.09 | Yes | No | 0.285 | -2.461 |
| B22 | No | 0.75 | No | Yes | 2.83 | -0.135 | Yes | No | 0.285 | -2.074 |
| B23 | No | 0.754 | No | Yes | 2.845 | 0.147 | Yes | No | 0.285 | -2.222 |
| B24 | No | 0.765 | No | Yes | 2.834 | -0.099 | Yes | No | 0.285 | -2.315 |
| B25 | No | 0.774 | No | Yes | 2.814 | -0.302 | No | No | 0.285 | -2.629 |
| B26 | No | 0.765 | No | Yes | 2.824 | -0.09 | Yes | No | 0.285 | -2.586 |
| B27 | No | 0.763 | No | Yes | 2.833 | -0.093 | No | No | 0.285 | -2.726 |
Result showed that all compounds showed no risk indication of skin sensitization, hERG I Inhibitory activity, AMES toxicity and hepato-toxicity. Every substance exhibited hERG II inhibitory action. Oral acute and chronic toxicity limits are 2.175 to 2.991 and -0.088 to 1.581, respectfully. Minnow toxicity of all compounds was found in the range of -0.026 to -0.504 and thus these molecules are anticipated to be a low risk of acute toxicity.
DISCUSSION
In 2018, there were anticipated to be 1,735,350 new instances of cancer reported, and 609,640 cancers related fatalities globally. Cancer is a second biggest cause of death. By 2025, there might be 19.3 million additional cases, an increase from the current level. As a result, there is still a pressing need for efficient chemotherapeutic drugs to the treatment of cancer with fewer side effects and combat multidrug resistance.24,25 Naturally occurring products have long been exploited as model molecules for the formation of new molecules improved pharmacological response for a variety of disorders, such as cancer. In this regards, podophyllotoxin is considered as a pioneer in the development of new chemical entities for the treatment of cancer.26–28
Several research teams have made significant synthetic efforts to increase the selectivity as well as DNA topo II inhibition. Earlier research on structure-activity relationship have suggested that the core structural components of the 3,4,5-trimethoxyphenyl fragment and the transfused methylenedioxyphenyl ring are important for the anticancer potential of podophyllotoxin derivatives.29–31
On account of these significant contributions of methylenedioxyphenyl moiety and the hybridization of two or more bioactive fragments may have complementary or increased effectiveness and lessen drug resistance issues. Due to their high capacity to produce DNA damage, topo II poisons are still widely used to treat cancer today. Thus, we have designed a series of methylenedioxyphenyl derivatives with other heterocyclic nucleus (pyrimidine, and piperazine) that would modify the drug skeleton to promote anticancer activity, reduce drug resistance problems as well as its severe toxicity.
The active site of topoisomerase IIα complexed with DNA and etoposide (PDB ID-5GWK) displayed a superior docking score in the examination of developed compounds using molecular docking. In all, ligand B18 achieved a best binding affinity of -11.3 and established good hydrogen bond interactions with SER 464 (Figure 3). The ligand B18 exhibited hydrophobic interactions with ASP463, MET766, ARG487, MET762, PRO803, GLY462, DG13, DC8, DT9, DA12, MET769, GLU461, GLY488, TYR805, and GLY760 is shown in Table 3. The top ranked ligand B18 considering docking score, hydrogen bond and hydrophobic interactions were selected for further studies. The co-crystal ligands were isolated and re-docked (Figure 3) into the topoisomerase II alpha binding pocket for validation purposes. The co-crystallized ligand achieved binding affinity of -10.2 and established hydrogen bond interactions with ASP463 and MET766. It exhibited hydrophobic interactions with ARG487, MET762, PRO803, GLY462, DG13, DC8, and DT9. Although, the majority of the compounds docked on the enzyme topoisomerase II produced better results than the standard molecule.
The physicochemical, pharmacokinetic and drug likeness characteristics of all the proposed compounds were calculated using the SwissADME and pkCSM software. Compounds with undesirable properties, particularly those with poor ADMET (absorption, distribution, metabolism, excretion, and toxicity) properties, should be avoided, have been frequently identified using the physicochemical properties of existing small organic pharmaceuticals and therapeutic prospects. Druglikeness rating can be used to exclude molecules from clinical trials that are anticipated to fail early on, which is important for raising the success rates and reducing the financial costs of drug development. Although experimental findings should back up the docking and ADME/Tox investigations, it is obvious from the results signify that compounds potential topoisomerase II inhibitors.
CONCLUSION
DNA topoisomerases play a key role in maintaining the topology of DNA in cells. Their function in cell development and proliferation, which can be abnormal and lead to the emergence of malignancies, has also been discussed. For finding new hit compounds, virtual screening techniques have proved a valuable resource. In this work, twenty seven hypothetically created molecules are docked using with Autodock vina programme to show how they interact with the active sites of DNA topo II, which has the PDB ID-5GWK. The docking investigations led to conclusion that some molecules from the designed series displayed superior docking score than the reference medication etoposide. These molecules have binding energies ranged from -10.2 to -11.3, and for etoposide binding energy is -10.2. These molecules exhibited potent interactions with human topoisomerase IIα enzyme; they bound to the DNA and the enzyme’s active site residues. Additionally, the SwissADME online tool’s predictions of all pharmacokinetic parameters were all within the acceptable range. We came to the conclusion that some of the best chemical structures had significant topo II inhibitory profile, favourable ADME/Tox parameters. These molecules may prove to be effective topoisomerase IIα inhibitors in the future. The synthesis of these compounds is taken up in laboratory.
References
- Wang JC. Cellular roles of DNA topoisomerases: A molecular perspective. Nat Rev Mol Cell Biol. 2002;3(6):430-40. [PubMed] | [CrossRef] | [Google Scholar]
- Cowell IG, Sondka Z, Smith K, Lee KC, Manville CM, Sidorczuk-Lesthuruge M, et al. Model for MLL translocations in the rapyrelated leukemia involving topoisomerase II β-mediated DNA strand breaks and gene proximity. Proc Natl Acad Sci U S A. 2012;109(23):8989-94. [PubMed] | [CrossRef] | [Google Scholar]
- Tiwari VK, Burger L, Nikoletopoulou V, Deogracias R, Thakurela S, Wirbelauer C, et al. Target genes of topoisomerase II β regulate neuronal survival and are defined by their chromatinstate. Proc Natl Acad Sci USA.. 2012;109(16):E934-43. [PubMed] | [CrossRef] | [Google Scholar]
- Pogorelčnik B, Perdih A, Solmajer T.. Recent advances in the development of catalytic inhibitors of human DNA topoisomerase IIα as novel anticancer agents. Curr Med Chem. 2013;20(5):694-709. [PubMed] | [CrossRef] | [Google Scholar]
- Larsen AK, Escargueil AE, Skladanowski A.. Catalytic topoisomerase II inhibitors in cancer therapy. Pharmacol Ther. 2003;99(2):167-81. [PubMed] | [CrossRef] | [Google Scholar]
- Farr CJ, Antoniou-Kourounioti M, Mimmack ML, Volkov A, Porter AC. The α isoform of topoisomerase II is required for hyper compaction of mitotic chromosomes in human cells. Nucleic Acids Res. 2014;42(7):4414-26. [PubMed] | [CrossRef] | [Google Scholar]
- D Arcy N, Gabrielli B.. Topoisomerase II inhibitors and poisons, and the influence of cell cycle check points. Curr Med Chem. 2017;24(15):1504-19. [PubMed] | [CrossRef] | [Google Scholar]
- Diniz EMLP, Poiani JGC, Taft CA, da Silva CHTP. Structure-based drug design, Molecular Dynamics and ADME/Tox to investigate proteinkinase anti-cancer agents. Curr Bioact Compd.. 2017;13(3):213-22. [CrossRef] | [Google Scholar]
- Verma P, Tiwari M, Tiwari V.. Array. J Biomol Struct Dyn.. 2018;36(5):1182-94. [PubMed] | [CrossRef] | [Google Scholar]
- Wendorff TJ, Schmidt BH, Heslop P, Austin CA, Berger JM. The structure of DNA-bound human topoisomerase II alpha: conformational mechanisms for coordinating inter-subunit interactions with DNA cleavage. J Mol Biol. 2012;424(3-4):109-24. [PubMed] | [CrossRef] | [Google Scholar]
- Wang YR, Chen SF, Wu CC, Liao YW, Lin TS, Liu KT, et al. Producing irreversible topoisomerase II-mediated DNA breaks by site-specific Pt(II)-methionine coordination chemistry. Nucleic Acids Res. 2017;45(18):10861-71. [CrossRef] | [Google Scholar]
- YİLMAZ S, ATAEİ S, YILDIZ İ. Molecular docking studies on some benzamide derivatives as topoisomerase inhibitors. J Fac Pharm Ankara. 2020;44(3):470-80. [CrossRef] | [Google Scholar]
- Ertan-Bolelli T, Bolelli K.. Discovery of new DNA topoisomerase II inhibitors using structure based virtual screening method. J Turk ChemSoc. 2019;6(1):71-8. [CrossRef] | [Google Scholar]
- Yu C, Hu J, Luyten W, Sun D, Jiang T. Identification of novel topoisomerase II alpha inhibitors by virtual screening, molecular docking, and bioassay. Chem Biol Drug Des.. 2022;99(1):92-102. [PubMed] | [CrossRef] | [Google Scholar]
- Seeliger D, de Groot BL. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. JComput Aid Mol Des. 2010;24(5):417-22. [PubMed] | [CrossRef] | [Google Scholar]
- Muegge I, Rarey M.. Small molecule docking and scoring. Rev Comput Chem. 2001;17:1-60. [CrossRef] | [Google Scholar]
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev.. 1997;23(1-3):3-26. [CrossRef] | [Google Scholar]
- Bkhaitan MM, Mirza AZ, Shamshad H, Ali HI. Identification of potent virtual leads and ADME prediction of isoxazolidine podophyllotoxin derivatives as topoisomerase II and tubulin inhibitors. J Mol Graph Model.. 2017;73:74-93. [PubMed] | [CrossRef] | [Google Scholar]
- Patil TD, Thakare SV. Array. Int J Pharm Pharm Sci.. 2012;4(4):201-4. [PubMed] | [CrossRef] | [Google Scholar]
- Muegge I, Heald SL, Brittelli D.. Simple selection criteria for drug-like chemical matter. JMedChem. 2001;44(12):1841-6. [PubMed] | [CrossRef] | [Google Scholar]
- Egan WJ, Merz KM, Baldwin JJ. Prediction of drug absorption using multivariate statistics. JMedChem. 2000;43(21):3867-77. [PubMed] | [CrossRef] | [Google Scholar]
- Nanjundaswamy S, Gurumallappa Hema MK, Karthik CS, Rajabathar JR, Arokiyaraj S. Synthesis, crystal structure, ADMET, molecular docking and dynamics simulation studies of thiophene-chalcone analogues. JMolStruct.. 2022;1247:131365 [CrossRef] | [Google Scholar]
- Gurumallappa G, Jayaprakash J, Puttaswamy AA, Sunderraj JH, Mallu P, Beeregowda KN, et al. 4-(Tert-butyl)-N,N-diethyl benzenesulfonamide: structural, absorption distribution metabolism excretion toxicity (ADMET) and molecular docking studies. Curr Chem Lett.. 2021;10:329-36. [CrossRef] | [Google Scholar]
- Khalil OM, Gedawy EM, El-Malah AA, Adly ME. Novel nalidixic acid derivatives targeting topoisomerase II enzyme; Design, synthesis, anticancer activity and effect on cell cycle profile. Bioorg Chem. 2019;83:262-76. [PubMed] | [CrossRef] | [Google Scholar]
- Hu W, Huang XS, Wu JF, Yang L, Zheng YT, Shen YM, et al. Discovery of novel topoisomerase II inhibitors by medicinal chemistry approaches. J Med Chem. 2018;61(20):8947-80. [PubMed] | [CrossRef] | [Google Scholar]
- Zhang X, Rakesh KP, Shantharam CS, Manukumar HM, Asiri AM, Marwani HM, et al. Podophyllotoxin derivatives as an excellent anti-cancer aspirant for future chemotherapy: A key current imminent needs. Bioorg Med Chem. 2018;26(2):340-55. [PubMed] | [CrossRef] | [Google Scholar]
- Vishnuvardhan MVPS, V SR, Chandrasekhar K, Lakshma Nayak V, Sayeed IB, Alarifi A, et al. Click chemistry-assisted synthesis of triazolo linked podophyllotoxin conjugates as tubulin polymerization inhibitors. Med Chem Comm. 2017;8(9):1817-23. [PubMed] | [CrossRef] | [Google Scholar]
- Hussain M, Khera RA, Iqbal J, Khalid M, Hanif MA. Phytochemicals: key to effective anti-cancer drugs. Mini Rev Org Chem. 2019;16(2):141-58. [CrossRef] | [Google Scholar]
- Reddy VG, Bonam SR, Reddy TS, Akunuri R, Naidu VGM, Nayak VL, et al. 4β-amidotriazole linked podophyllotoxin congeners: DNA topoisomerase-IIα inhibition and potential anti-canceragentsforprostatecancer. Eur JMed Chem.. 2018;144:595-611. [PubMed] | [CrossRef] | [Google Scholar]
- Karki R, Park C, Jun KY, Jee JG, Lee JH, Thapa P, et al. Synthesis, antitumor activity, and structure-activity relationship study of trihydroxylated 2, 4, 6-triphenyl pyridines as potent and selective topoisomerase II inhibitors. Eur J Med Chem. 2014;84:555-65. [PubMed] | [CrossRef] | [Google Scholar]
- Li Z, Luo M, Cai B, Haroon-Ur-Rashid , Huang M, Jiang J, et al. Design, synthesis, biological evaluation and structure–activity relationship of sophoridine derivatives bearing pyrrole or indole scaffold as potential antitumor agents. Eur J Med Chem. 2018;157:665-82. [PubMed] | [CrossRef] | [Google Scholar]