Corresponding author.
Author Notes
Copyright and License information
ABSTRACT
Background
Cancer stands as a significant global health concern, leading to considerable patient suffering and death. The prognosis of patients is intricately connected to the metastatic extent of the disease to aloof sites, where metastatic conditions contribute substantially to cancer-related mortality.
Materials and Methods
Despite progress in the field, our understanding of the mechanisms governing cancer metastasis and its establishment at secondary sites remains limited. The TME (Tumor Microenvironment) controls vital tumor survival and advertising functions. Interactions within the TME, involving both cellular and structural apparatuses, enable cancer cells to acquire invasiveness and disseminate through a complex and multistep metastatic cascade from the crucial site to distant sites.
Results
In this review article, we have discussed tumor invasion and metastasis as well as the cellular and molecular events tangled in this complex process, like EMT, Cell adhesion, Angiogenesis and lymphangiogenesis, Intravasation and Extravasation, signaling pathways that govern tumor invasion and metastasis.
Conclusion
The part of stromal cells and the extracellular matrix in facilitating invasion and metastasis, current and emerging therapeutic approaches aimed at inhibiting tumor invasion and metastasis.
INTRODUCTION
Cancer cells must separate from the original tumor, move through surrounding tissue, enter and pass over the vasculature, and then survive and multiply in distant organs. This is a complicated, multi-step process known as metastasis.1 Carcinomas present an additional difficulty since the adult epithelial cells that give birth to them are typically nonmotile and polarized. Therefore, a metastatic state marks a dramatic departure from homeostasis. Nevertheless, during tissue repair and development, epithelial tissues are very dynamic and motile, and in the embryonic and pathological phases, epithelial cells can obtain mesenchymal molecular characteristics.2 We use the term “metastasis” to describe the foundation of secondary tumors in a body section distant from the initial primary cancer. Even though metastasis is the primary factor contributing to cancer therapy failure and death, little is known about it. Large amounts of cancer cells are squared into the bloodstream daily in cancer patients; however, melanoma investigation in animal models indicates that less than 0.1% of tumor cells metastasize.3 Cancer cells must travel from their original place, circulate through the bloodstream, withstand pressure in blood vessels, adapt to novel biological conditions in a secondary location, and avoid lethal interactions by immune cells for metastases to occur.1,4 According to Hanahan and Weinberg,3 one of the characteristics of cancer is” Inducing invasion and metastatic processes.” A key component of cancer malignancy continues to be the invasion of neighboring tissue and the seeding of distant places to create metastases. After all, for more than 90% of cancer patients, metastasis is the prominent origin of mortality.3 In this review article, we have discussed tumor invasion and metastasis, their role in cancer progression, and recent advancements and focus insights that give to influential this mark of cancer.
Epithelial-Mesenchymal Transition (EMT)
EMT trans differentiation, altered epithelial cells acquire the capacity to attack, withstand stress, and increase.3 In addition to being firmly attached to the surrounding Extracellular Matrix (ECM) and one another, epithelial cells are immotile.5 Epithelial-Mesenchymal Transition (EMT) regulates the revocable biochemical changes that allow a particular epithelial cell to acquire a mesenchymal phenotype and endows epithelial-mesenchymal plasticity on epithelial cells,2 which is essential for the advancement and spread of cancer. However, not every cell that grows from the original tumor location aids in the metastatic process. Asparagine synthetase is a metabolic enzyme connected with the development of metastases, according to research on the factors influencing metastatic probable in a mouse model of breast cancer.6 Reducing asparagine levels by food restriction or ρ-asparaginase therapy inhibited the spread of metastatic disease. Therefore, the availability of asparagine encouraged EMT.6 In similarity to a process that entails a binary optimal between full-epithelial and full-mesenchymal phenotypes, it is now widely acknowledged that the EMT program is a range of transitional stages between the epithelial and mesenchymal phenotypes.7 Many growth factors and signaling pathways control the change from one state to another. In initial tumor cells, spontaneous EMT alternates between intermediate stages with multiple traits related to invasion, metastasis, and differentiation.3 Tumor cells that exhibit mesenchymal and epithelial phenotypes are more efficient in metastatic formation, circulation, and secondary site colonization.8 Furthermore, single-cell RNA sequencing, chromatin, and transcriptional analyses demonstrate that the different phases have unique chromatin landscapes, cellular features, and gene expression profiles controlled by shared and individual transcription factors and signaling cascades.
The different stages (EMT) interact with other stromal cells and are located in different microenvironments.8 For instance; the best marked mesenchymal phenotype metastatic cells grow proximate inflammatory and endothelial cells. These tumor cells discharge huge amounts of chemokines and proteins to draw in immune cells and trigger angiogenesis, which aids in forming a distinct inflammatory and highly vascularized niche.8 Through fibronectin alignment, cancer-associated fibroblasts have also been demonstrated to drive and direct the migration of cancer cells.9 Further factors that initiate the EMT pathway in cancer cells are hypoxia, metabolic stresses, and matrix stiffness.3,10 Transcription factors that are designed to activate mesenchymal genes and suppress epithelial genes are frequently responsible for driving transitioning.3 In addition, posttranslational and epigenetic modulators are essential for regulating the EMT process.7 Whether EMT plays a significant part in chemotherapy resistance and cancer metastasis has recently been hotly debated.3,11–13 Studies on pancreatic and lung malignancies indicate that although EMT may not be necessary for metastasis, it does play a role in chemo resistance.11,12 However, more data is required to fully and unambiguously explain how EMT contributes to the development of cancer and the metastatic process.
The opposing progression, known as the Mesenchymal-Epithelial Transition (MET), is essential for the advancement of metastatic disease, even if EMT may be necessary for the start of metastasis. In bone metastasis, WNT and MET activation in cancer cells is triggered by E-selectin in the bone vasculature, which propels the creation of metastatic tumors (Tables 1 and 2).14
| Properties | EMT | MET | References |
|---|---|---|---|
| Morphology | Polygonal-morphology. | Spindle-like morphology. | 2 |
| Polarity | Apical-based polarity. | Anterior-posterior polarity. | 15 |
| Cytoskeleton | Expresses cytokeratins. | Expresses vimentin. | 16 |
| Migratory potential | Limited migratory potential. | Strong migratory potential. | 16 |
| Junctions | Tight junctions, Adherens junctions. | No junctions | 16 |
| Markers | E-cadherin, Claudin, Occludin, Desmoglein, Desmocollin and Cytokeratins. | N-cadherin, Vimentin, Fibronectin, Snail/2, Fibroblast-specific protein and Smooth muscle actin. | 17 |
Difference between EMT and MET based on their properties.
| miRNAs | miRNAs inhibiting EMT | miRNAs promoting EMT | References |
|---|---|---|---|
| miR-10b | ☒ |  | 18–20 |
| miR-21 | ☒ | | 21–23 |
| miR-155 | ☒ | | 23 |
| miR-9 | ☒ | | 24,25 |
| miR-29a | ☒ | | 26 |
| miR-103/107 | ☒ | | 26 |
| miR-5003-3p | ☒ | | 27 |
| miR-181b-3p | ☒ | | 26 |
| miR-221/222 | ☒ | | 26 |
| miR-373 | ☒ | | 26 |
| miR-100 | ☒ | | 28 |
| miR-200 family | | ☒ | 29 |
| miR-34 family | | ☒ | 30,31 |
| miR-497 | | ☒ | 32 |
| miR-125b | | ☒ | 33 |
| miR-206 | | ☒ | 26 |
| miR-30a | | ☒ | 26 |
| miR-138 | | ☒ | 34 |
| miR-195 | | ☒ | 26 |
| miR-143 | | ☒ | 35 |
| miR-153 | | ☒ | 36 |
| miR-300 | | ☒ | 37 |
miRNAs inhibiting and promoting EMT.
Cell adhesion in normal tissues and the role of adhesion molecules in movement
Of the adhesion receptor classes investigated, most are integrins and cadherins. Cadherins mediate homotypic cell-to-cell adhesions, while integrins mediate adhesions between cells and their extracellular matrix. Cadherins and integrins are regarded as closely connected components of a broad sticky network due to their diverse interactions at various levels.38,39
The transmembrane proteins known as cadherins, which are dependent on calcium, are essential for the formation of junctional adhesions, which hold cells inside tissues together, preserve intracellular cohesion, and maintain tissue architecture. These proteins are necessary for tissue makeover, morphogenesis, and tissue barrier maintenance.39 One signal sequence, a protein precursor, a single transmembrane domain, and five ectodomains are commonly included in the structure of cadherins. Four of the ectodomains are associated with the extracellular cadherin domain, a protein module of the immunoglobulin-like fold that binds to calcium ions to aid in correct folding. Four conserved cysteines and an N-terminal extracellular domain are present in the fifth domain. A protein is considered a cadherin family member if it has one or more extracellular cadherin domains. Several isoforms of the cadherin superfamily are distributed differently depending on the tissue.39,40
It has been demonstrated that a subset of cadherins, known as classical cadherins, mediates homotypic adhesive contacts. E- and N-cadherin were the first conventional cadherins found in vertebrates. For classical cadherins to function as adhesives, proteases must remove the pro-domain.39 Furthermore, these proteins form an adhesive dimer interface in which a twofold symmetric connection is produced by switching the N-terminal domains of paired extracellular cadherin.1 When calcium is present, cadherins from adjacent cells preferentially create these strand-swapped interfaces. Densely packed junctional structures are produced when apposed adherent cells assemble cadherins.39 Integrins are the primary grade of receptors implicated in both homotypic and heterotypic adhesive events. These proteins are members of a vast and intricate family of trans-membrane glycoproteins that transfer signals both inside-out and outside-in. These proteins are structurally heterodimeric transmembrane receptors that are created when two of these trans-membrane glycoproteins associate non-covalently. These two subunits are designated as α and β, and it is currently known that eight β subunits and eight α subunits of integrin heterodimerize to form 24 distinct integrins.41 One transmembrane domain, ectodomain, and a brief cytoplasmic tail domain comprise the subunits α and β. Their extracellular domains consist of globular ligand-binding head regions and extended stalks that bind primarily to other glycoproteins and components of connective tissue, including collagen, laminins, and fibronectin. The adhesion location of integrin β subunits depends on metal ions and can bind, for instance, to magnesium ions. Integrin ligand interaction causes conformational alterations that result in an exposed and energetic integrin conformation by altering the metal ion-dependent adhesion location coordination through magnesium ions.39 In cell-matrix adhesions, most integrins are connected to the actin cytoskeleton using cytoskeletal linker proteins, like talin, paxillin, and vinculin. Activating downstream pathways requires these interactions between integrins and the actin cytoskeleton. Integrins thus serve as a conduit between the external habitat and motility-related cellular responses, including immune cell trafficking, hemostasis, and cancer cell migration.42 Integrin is linked to cell propagation and anchorage-dependent survival through various growth factor response pathways that rely on integrin-mediated adherence to the extracellular matrix or integrin-dependent intracellular signaling.39 The extracellular domains of immunoglobulin-like cell adhesion molecules, or Ig-CAMs, are heavily glycosylated and comprise varying numbers of immunoglobulin-like loops.39 Ig-CAM’s extracellular domain can be connected to a transmembrane domain or anchored in the membrane by glycophosphatidylinositol anchors. Homotypic contacts can mediate cell-to-cell adhesion between Ig-CAMs, whereas these proteins’ cytoplasmic tails can interact with cytoskeletal proteins. The T-cell receptor complex and key histocompatibility complex class I and II molecules are the most well-known members of this superfamily. ICAM, VCAM, MadCAM-1, and ALCAM are additional components crucial to leukocyte trafficking. An additional class of adhesion molecules associated with immune activity is called selectins. Selectins bind to carbohydrates in a Calcium (Ca) dependent way to mediate cell-cell adhesions. The leukocyte rolling process, which starts the immune cell’s migration via the blood artery wall and into the neighboring tissue, is brought on by these transmembrane proteins.39 All of the chemicals mentioned above have specific functions in context-dependent adhesion between cells and the extracellular matrix. However, adhesion molecules have a functional diversity that comes from their capacity to transduce signals from the environment and initiate intracellular answers in addition to outside-in transmission.
Although integrins are essential for single-cell migration, dependent on the total loss of adherens junctions, which E-cadherin facilitates, integrins are also sensitive to external stimuli and pressures that cause movement. Interacting with the extracellular matrix otherwise intracellular proteins, which modify integrin binding affinity, impact clustering, and attract cytoskeletal linker proteins, integrins undergo conformational modifications that enable them to carry out their diverse activities.43 While coordinated formation and breakdown of these adherent structures make forces of cellular migration, these alterations reshape nascent or focal adhesions and produce tension.39,3 Trafficking of immune cells is one of the many physiological processes that depend on single-cell migration and invasion. An additional mechanism of collective cell movement in morphogenesis and wound healing has also developed. During this process, cell assemblies move in conjunction while the cell-cell junctions hold firm, enabling nearby cells to stick to one another. Homotypic cadherin interactions between group members maintain adherens junctions during collective migration.39
Igs L1CAM, NCAM, and ALCAM are other adhesion molecule family members that can assist in this role.39 Because they can bind extracellular matrix deposits between cells, integrins also contribute to collective adhesion by supporting cell cohesion.39 Different forms of group migration, such as sheet migration and the movement of cellular strands and clusters, are caused by the variability of adhesion molecules and signaling contexts, which causes cell-cell junctions to become flexible.13
Angiogenesis and lymphangiogenesis
Patients rarely die from primary carcinomas (unless in cases of liver and lung cancer). Instead, metastasis-related consequences account for most carcinoma-related deaths. Joseph-Claude- Anthelme Recamier was the one who originally invented the term metastasis.44 Furthermore, a single organ may give rise to hundreds of metastatic colonies, and these colonies’ unchecked proliferation may cause tumors to replace normal tissue in vital organs like the bone marrow, lung, liver, and brain.44 Sarcomas seldom metastasize, in contrast to carcinomas and melanomas. Small avascular dormant lesions also do not spread. Since metastasis typically occurs later in a carcinoma, surgical excision can cure tiny tumors discovered early on. A series of connected, sequential rate-limiting processes are referred to as the metastatic process.44 For most malignant neoplasms, the sequence of steps in metastasis is identical, although the path and final destination can differ: (1) The formation of Angiogenesis, a vascular or lymphatic network, must occur in cancer cells.44 Neo vessels give the tumor the nutrition it needs to thrive and a way out, allowing the tumor cells to escape into the bloodstream. Generally speaking, the likelihood of a tumor cell escaping increases with tumor size and vascular density. (2) Intravasation: To reach the bloodstream, malignant tumor cells need to separate from their surrounding cells and matrix, penetrate the capillary cellar membrane, and move across the endothelium wall. It is believed that the mesenchymal phenotypic change facilitates this migration mechanism. Alternatively, the mixture of cells and fluid that enters lymphatic capillaries may pick up unattached tumor cells. Tumor cells may enter the circulatory system through either pathway because all lymphatic fluid is reused into the venous system. (3) Circulation survival: Tumor cells cannot survive amid the shear stress created by the pulsatile blood flow and maximum serum conc. (concentration). During the first 24 hr, most circulating tumor cells will perish through attrition or be directly cytotoxic and lysed by Natural Killer (NK) cells during immunosurveillance.45 In the early hours following intravasation, platelets shield tumor cells, and TGFβ produced from platelets may promote tumor cell extravasation through EMT pathways.44 (4) Extravasation: Tumor cells are believed to extravasate in distant organs via transmigrating across inter-endothelial junctions. According to recent research, cellular pathways can also be used for lymphocyte (and potentially tumor cell) diapedesis.44 Even when tumor cells enter the bloodstream through venules, they frequently go to arterioles where the tissue’s oxygenation level is higher. Metastatic patterning is determined by interactions between the host and tumor cell rather than by the cancer cells lodging in the first vascular bed they come across. (5) Development of secondary tumors: Successful Metastases are not always the consequence of extravasation. Once removed from circulation, most solitary cancer cells either undergo apoptosis, are destroyed by immune cells, or stay dormant.44 Malignant tumor cells must multiply and go through Angiogenesis once more to become a clinically significant secondary tumor, forming macrometastases. Tumors that have spread to other secondary sites and even back into the original tumor can re-metastasize.44 The correlation between tumor microvessel density, low survival in almost entirely arrangements of malignancy, and increased risk for metastasis is one of the most precise lines of suggestion connecting Angiogenesis and metastases. Lung micrometastases cannot grow unless Angiogenesis occurs (Figure 1). While some studies claim that coopting normal arteries is a mechanism for metastasis vascularization, others imply that EPC generated from bone marrow contributes to the early angiogenic stages of metastatic development.44 Contrary to popular belief, Folkman et al. demonstrated that removing a primary tumor from a mouse could encourage the formation of dormant metastases.44 Angiostatin, an endogenous angiogenesis compounds (inhibitor) released systemically by way of the principal tumor, was lost, which accounted for the angiogenic flip in the metastatic site. Likewise, the promotion of tumor vascularization and metastasis is observed with the deletion of additional endogenous angiogenesis suppressors.44 These findings further support the relationship between Angiogenesis and metastasis. Remarkably, pericytes are involved in metastasis as well. Pericytes are crucial for the spread of tumor cells because they regulate the patency of capillary lumens. MMP9 mutant mice have reduced metastasis, collapsing vascular architecture, and a lack of pericytes.46
In addition to extravasation, Circulating Tumor Cells (CTCs) can settle in remote organ microvessels and initiate intraluminal growth. Eventually, these cells might break through the vessel wall and reach the tissue beneath.44 Because they denote a particular stage in the metastatic process, CTCs also stand for “metastatic intermediates.” Because of this, these cells are increasingly highly prized in preclinical and clinical trials as proxy indicators of therapeutic success.47 To put it another way, fewer CTCs should be discharged into the bloodstream if a medication is successful in decreasing a tumor. Drugs that lower CTC levels may be applicants for clinical development, as great CTC numbers are correlated by way of poor clinical outcomes.
The tropism of some cancers for particular organs is explained by the “seed and soil” theory, which was proposed by Paget and made famous by Fidler.44 This could be because some tumors have receptors for particular growth factors produced specially from a specific organ. Colon cancer cells, for example, that expressed the cMET receptor migrated to the liver and secreted HGF.44 Alternatively, tumor cells may recognize “zipcodes” or surface markers on tissue-specific vascular beds. It has been observed that some primary tumors build a pre-metastatic niche in the secondary location before the entrance of tumor cells. In contrast, the systemic induction of thrombospondin-1 expression of lung stromal cells was suppressed by primary tumor cells secreting prosaposin, which prevented metastatic colonization.44
Lymphangiogenesis
Veins give rise to lymphatic channels throughout the middle of gestation (E9-9.5 in mice). Nonetheless, the lymphatic system differs significantly from the blood-vascular system in structure and function in an adult.44 The body’s vascular system is a closed circulatory system that pumps blood via venules, veins, arteries, and arterioles to supply oxygen and nutrients to every cell. Conversely, lymphatic tubes are part of a unidirectional fluid recycling system. Lymphatic capillaries absorb fluid (and cells), and liquid propulsion is directed toward Lymph Nodes (LN) via lymphatic collecting ducts, including valves. Even though fluid may pass through multiple LNs for filtering, lymphatic fluid ultimately returns to the venous system.44 Lymphatic capillaries differ architecturally from vascular capillaries at the microvessel level. The endothelium lining lymphatic capillaries is weak, lacking pericyte covering and lacking a basement membrane entirely. When the pressure in the interstitial space increases, anchoring filaments open tiny gaps at button junctions between adjacent Lymphatic EC (LEC)44 through improving drainage, these intercellular LEC gaps are crucial for controlling fluid dynamics and preventing tissue edema.
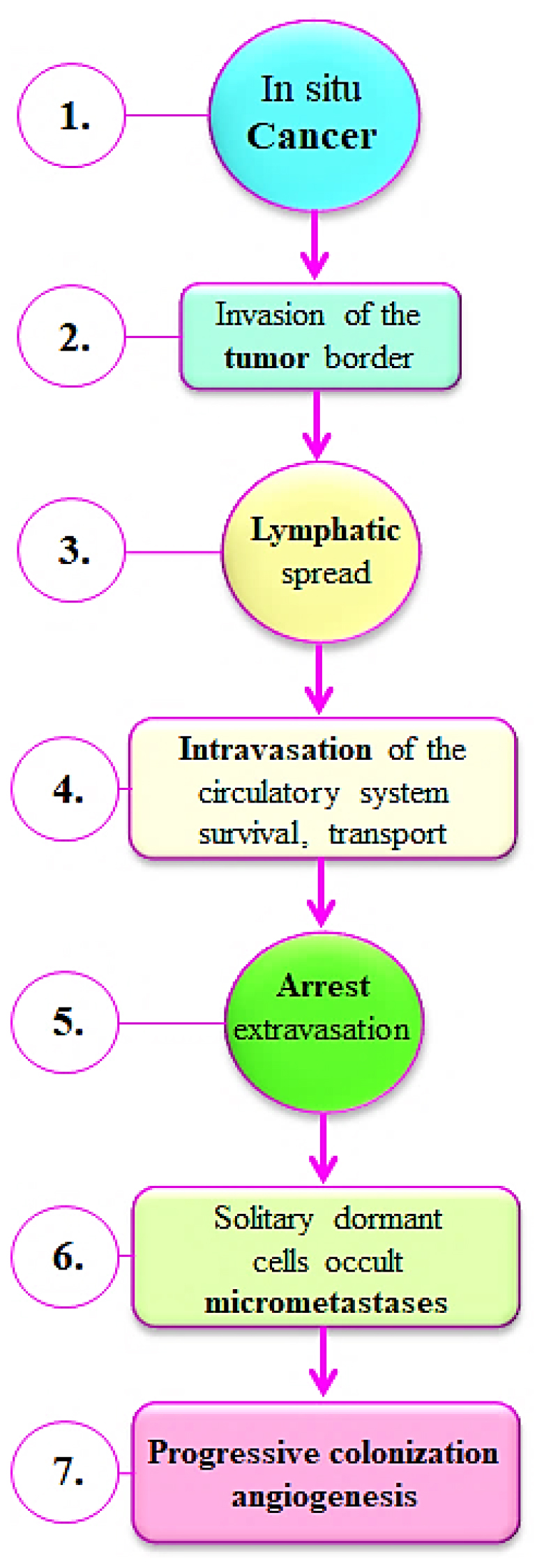
Figure 1:
This flowchart preserves the orderly flow and offers a thorough explanation of the many phases, starting with the early stages of in situ cancer growth followed by Invasion of the tumor edge, Lymphatic spread, Intravasation of the circulatory system survival, transport, Arrest extravasation, Solitary dormant cells occult micrometastases and ending with the later phases of increasing colonization and angiogenesis.
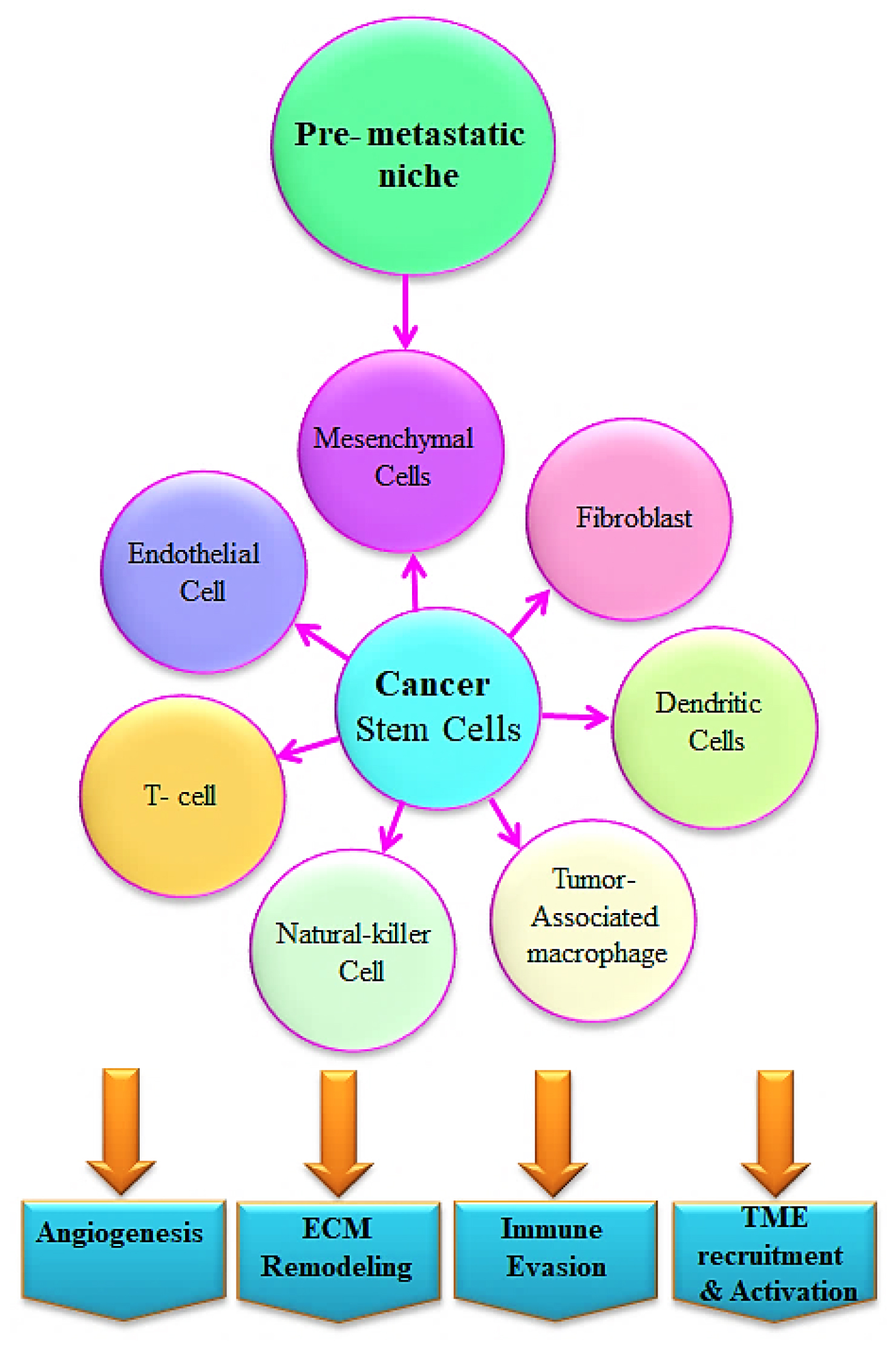
Figure 2:
Interaction of CSC through the tumor microenvironment- The primary cause of tumor development and metastasis is CSCs. The interaction between CSCs and the tumor microenvironment, which includes T cells, natural killer cells, dendritic cells, Tumor-Associated Macrophages (TAMs), mesenchymal stem cells, fibroblasts, and endothelial cells, is one of the key factors impelling the development of cancer and resistance to drug treatments. Through release of growth factors, cytokines, and Extracellular Vesicles (EVs), these cells interact with CSCs to promote tumor development and immune evasion.
Additionally, they provide access to the LN for immune cell infiltration, including dendritic and Langerhans cells. Above all, these apertures provide tumor cells with both passive and direct access to the lymphatic system. The LN serves as a filter, protecting the host at first, but it can also serve as a hub or breeding ground for tumor cell pooling, which can later move to other locations. Because certain tumor cells require the “key” (CCR8) to unlock the “gate” (CCL1 expression by LEC) at the LN sinus, access to the lymphatic system does not guarantee that tumor cells will reach the LN.44 There are reports of metastases to regional Lymph Nodes (LNs) from malignant carcinomas of almost any kind.44 Certain malignancies appear to spread in a metachronous manner, with a tumor in the sentinel (draining) LN before metastases in distant organs. In contrast, other cancers spread simultaneously via the vascular and lymphatic systems. A lymphadenectomy may be beneficial in these consecutive cases. Tumor classification using TNM staging is predicated on the idea that supreme tumors spread consecutively, starting with a crucial tumor, moving on to lymph node metastasis, and ending with distant metastases.44
Figure 3:
The side effects of Chemotherapy on the Human Body.
Chemotactic proteins secreted by tumor-associated LEC can draw malignant tumor cells.44 Similar to tumor angiogenesis, tumor lymphangiogenesis is a growth factor-mediated process that produces hyperplastic capillaries that are poorly formed and increase the microvascular density and area available for tumor cell escape. Tumor lymphangiogenesis, however, varies from angiogenesis in that it primarily involves the enlargement of the peritumoral lymphatic veins and the emergence of a small number of lymphatic capillaries inside the tumor. The lymphatic vessel area metric was a more reliable indicator of metastasis in histological human melanoma patient samples than the gold standard tumor thickness.44 Increased LN metastasis and stimulation of tumor-associated lymphangiogenesis within and around the tumor periphery were observed in tumors designed to overexpress the lymphangiogenic factor, VEGFC.44 On the other hand, LN metastasis remained unaffected when VEGFC was silenced in a different paradigm that only affected the intratumoral LEC, indicating that peritumoral lymphatic capillaries are adequate for LN metastasis.44
Tumor cells that have made their way into the Lymph Node (LN) behave like a second tumor and can either infiltrate local blood vessels or continue their journey through the lymph to reach the bloodstream finally. It is now understood that the lymphatic system is a substantial source of CTCs in the blood. In most malignancies, lymph node metastasis is associated by a poor clinical result and an elevated risk of distant metastases. The level to which LN metastases themselves populate metastases in nearby or distant non-lymphatic organs remains an open subject.44
Extravasation of tumor cells, although tumor cells employ a variety of tactics to endure the harsh intravascular environment, their capacity to spread across the surrounding tissue quickly ultimately determines whether or not they can metastasize.48 Tumor cell extravasation is thought to occur by endothelial adhesion, endothelial barrier modification, and transendothelial migration to the underlying tissues. Tumor cells can rove or multiply inside blood arteries and then extravasate,48 or they can openly transmigrate the endothelium as single cells and invade the tissues after being arrested, primarily in tiny capillaries. Paracellular migration, in which tumor cells move between two endothelial cells, appears to be the most common mode of extravasation.48 This process involves cellular rearrangements and the destruction of inter-endothelial cell-cell connections. Studies conducted in vitro have demonstrated that, in rare instances, tumor cells can penetrate individual cell bodies to move through the endothelium. This process is known as transcellular migration.48
They can directly aid the extravasation of tumor cells. For instance, it has been demonstrated that breast cancer cells express angiopoietin-like 4 (ANGPTL4) or its C-terminal fibrinogen-like domain (cANGPTL4), which impedes vascular endothelial tight and adheres junctions and promotes the extravasation and dissemination of tumor cells.48 To promote metastasis, it has been demonstrated that the C-terminal domain of ANGPTL4 directly interacts through integrin α5β1, VE-cadherin, and claudin-5 in a temporally serial way.48 Research has shown that the expression of Epidermal Growth Factor (EGF) receptor ligands, including Epiregulin (EREG) and Heparin-Binding EGF-like Growth Factor (HBEGF), along with the prostaglandin-synthesizing enzyme cyclooxygenase 2 (COX2), and the matrix-remodeling metalloproteinases MMP1 and MMP2, contributes to the promotion of breast cancer cell extravasation.48
The fundamental mechanisms are yet unknown, though. Osteonectin (SPARC) production increases the extravasation and metastasis of melanoma cells by binding to VCAM-1, promoting actin remodeling, and opening endothelial junctions.49 Another study demonstrated that tumor cells promote extravasation and metastasis through causing endothelial cells to undergo necroptosis, or programmed necrosis.50 Endothelial cell death occurs when the membrane-bound Amyloid Precursor Protein (APP) on tumor cells interacts to the endothelial Death Receptor 6 (DR6). This creates holes that allow tumor cells to invade. In adding to this straight pathway across the endothelium, extravasation can be facilitated by the endothelial barrier opening thru producing DAMPs (damage-associated molecular patterns) from necroptotic endothelial cells, such as ATP or HMGB1.48 Effective extravasation necessitates assistance from immune system cells that promote metastasis in adding to straight interactions between tumor cells and endothelial cells. DAMPs may collaborate by immune cells to facilitate extravasation. Epidemiological evidence suggests that elevated APP expression in tumor cells is linked to a greater likelihood of metastasis development and a less favorable outcome.51
Lymphatic intravasation vs hematogenous intravasation
Tumor cells can enter blood vessels otherwise lymphatic vessels to enter the circulation, with hematogenous (blood vessel) penetration accounting for most cases. The shear stress of blood flow only may be sufficient to kill off numerous cells entering the bloodstream due to the highly inefficient intravasation process. To cause distant metastasis, the cancer cells must bypass the immune system once they are in the bloodstream and implant in a secondary site.
The type of tumor, the configuration of the blood vessels, and the circumstances in the tumor microenvironment can all influence whether hematogenous intravasation is an active or passive process. The tumor cells themselves undergo a variety of alterations during intravasation. It has been observed that tumor cells that detect a gradient of chemo attractants can alter their cytoskeletal activity. Additionally, sign of the overexpression of integrins and other adhesion molecules to promote tumor cell attachment to endothelial cells has been reported.52 Tumor cells can also enter the bloodstream through lymphatic intravasation or enter into the lymphatics. Tumor cells can indirectly reach the venous blood supply through lymph capillaries that eventually leak into the blood after converging on the significant thoracic duct. However, during the journey, the tumor cells come across several lymph nodes, frequently the first locations where metastases of various malignancies are seen. The nearest sentinel lymph nodes to the crucial tumor site are removed and biopsied to look for any initial indicators of metastasis. Many factors determine whether a tumor cell invades the body by the lymphatic or hematogenous route.52 Tumor cell accessibility to arteries could be one apparent factor. Tumor angiogenesis forms a network of microvasculature that can subsequently access tumor cells. Tumors have also been shown to undergo lymphangiogenesis; however, it’s possible that the lymph vessels inside the tumor will not perform as effectively as those that grow outside the tumor tissue.52 The comparative proportions of blood vessels and lymphatics generated through the cancer may determine the quantity of hematogenous versus lymphatic intravasation. The modes of intravasation of blood arteries and lymphatics differ because of their structural distinctions. Lymphatics lack the taut endothelial connections found in blood vessels structurally. Tumor cells can enter lymphatics rather quickly, but their capacity to spread through connective tissue that stands in their way may restrict their ability to enter the lymphatics. Furthermore, lymphatic channels flow significantly less than blood vessels, reducing shear stress and facilitating lymphatic survival.52 Therefore, the capacity of tumor cells to cross connective tissue barriers may be reflected in the presence of metastases in nearby lymph nodes.
Nevertheless, breaking through the blood endothelial cell barrier can call for skills beyond straightforward invasion. As a result, there is disagreement over the lymphatic route’s significance in the development of distant metastases. The existence of a chain of lymph nodes carrying tumor cells that travel to the central thoracic duct does not strictly correlate with the development of distant metastases.52 Direct intravasation into the blood supply, either within the main tumor or maybe in the lymph nodes, is most likely the first stage in the development of distant metastases.
LKB1/AMPK signaling controls the invasion and migration of malignant cells
In addition to being a well-known tumor suppressor, LKB1 kinases the tumor energy metabolism sensor AMPK upstream. Tumor cells lacking LKB1 exhibit an upregulation of SNAIL, an EMT transcriptional factor.53 Likewise, ZEB1 was observed to be enhanced in lung cancer cells depleted of LKB1, immortalized epithelial cells, and mesenchymal marker protein expression, cell viability, and invasiveness.53 The results of double knocking down the phosphatase and tensin homolog removed in chromosome ten (PTEN) and LKB1 in mouse bladder tissues showed increased tumor initiation and cell proliferation, EMT, upregulation of SNAIL, vimentin nuclear localization, and decreased expression of tight junction protein Zonal Occludin-1 (ZO-1) and E-cadherin.53 These findings suggest that LKB1 is essential for the inhibition of EMT during the development of tumors.
A preserved downstream kinase of LKB1, AMPK has dual regulatory actions on cancer cell invasion and suppressing tumor cell proliferation and energy metabolism. According to specific research, AMPK triggers cell migration. Lysophosphatidic Acid (LPA) can activate AMPK and encourage the spread of ovarian cancer.53 According to Park et al., AMPK silencing eliminated anthocyanins’ capacity to prevent hepatocarcinoma cells from migrating. More recent research, however, emphasizes AMPK’s capacity to control tumor invasion and growth adversely. It has been observed that the AMPK activator metformin inhibits melanoma metastasis and EMT in an AMPK/p53-dependent way.53 Metformin has been seen to impede the levels of MMP-2, MMP-9 and the movement and proliferation of human umbilical vein endothelial cells. Human chondrosarcoma cells can migrate and express MMP-2 when Chemokine Ligand 3 (CCL-3) is present; however, this migration is stopped when AMPK activation occurs. According to Kim et al., berberine suppressed human melanoma cell motility by significantly raising AMPK phosphorylation while lowering ERK activity and Cytochrome c Oxidase subunit II (COX-2) expression.53 These findings imply that AMPK is crucial for the invasion and migration of tumors.
Through a variety of signaling pathways, the LKB1/AMPK system controls the invasion and migration of cancer cells.
The complicated process of tumor cell invasion and metastasis is caused by interactions and crosstalk across several pathways. With the help of several different signaling pathways, including TGF-β, mTOR, NF-κB, AKT, and forkhead box O3 (Foxo3a), the LKB1/AMPK pathway controls invasion and metastasis. AICAR has been shown to activate AMPK to reduce AKT/FOXO3 signaling, which prevents EMT and reverses the mesenchymal features of tumor cells.53 AICAR and A769662 reduced migration by activating AMPK and down regulating mTOR and S6K1 levels in PC3 and PC3M cells.53 Recent findings suggest that LKB1/AMPK suppresses tumor invasion and migration by down regulating TGF-β signaling downstream components such as MMPs and Snail.53 It was shown that in HK-2 cells, AMPK suppressed TGF-β-induced EMT.53 Furthermore, it was discovered that AICAR, metformin, and adiponectin activated AMPK; however, TGF-β-induced fibrosis was prevented by down regulating the expression of α-SMA and Collagen type I α 1 (COL1A) in hepatic stellate cells.53 The fundamental activation mechanism is AMPK-mediated proteasomal degradation of p300. Furthermore, TGF-β functions were reduced when metformin was administered to breast cancer cells. Conversely, there was tumor growth enhanced by RNA interference-silencing Lipopolysaccharide-Induced TNF-α Factor (LITAF), an AMPK downstream effector. It has also been demonstrated that metformin suppresses the AKT and ERK signaling pathways, which stop the course of the cell cycle, impede propagation, and trigger apoptosis in breast cancer cells. On the other hand, prostate cancer cells that had their AMPK inhibition increased in number and migration, increased S6K1 and Insulin-like Growth Factor 1 (IGF-1), and decreased p53 and p21.53
Via several different signaling pathways, AMPK can indirectly affect the development of tumors. By blocking the AMPK-NF-κB pathway, capsaicin prevented cholangiocarcinoma cells from migrating and invading, resulting in lower MMP-9 production. It has been discovered that the AMPK/JAK/STATA3 and AMPK/ GSK3β/β-catenin pathways are implicated in the invasion and metastasis of hepatocarcinoma and endometrial cancer cells.53 Members of the AMPK family, Novel (nua) Kinase family 1 (NUAK1), and ARK5 regulate the AKT and NF-κB signaling pathways, which are critical for cancer cell motility and invasion.30
The tumor microenvironment and its influence
It is widely acknowledged that immune cells, by either tumor infiltration or systemic environment modulation, play critical roles in the characteristics of cancer cells at various periods of the invasion-metastasis pour.53 Every stage of this deadly process exposes cancer cells to immune system responses intended to stop their proliferation.54 The key mediators of these anti-tumor effects are CD8+ T cells and Natural Killer (NK) cells. It has been demonstrated that these cells limit the metastatic spread of tumor cells, while their removal increases metastasis without influencing the growth of the original tumor.54 But when tumors evolve, cancer cells learn how to boost their metastatic effectiveness by evading immune monitoring and triggering systemic reactions through immune cell subtypes such as myeloid cells (Figure 2).54
Macrophages, which are produced from HSC (hematopoietic stem cells) in the bone marrow and are regarded as “professional” APCs (antigen-presenting cells), are the primary kind of myeloid cells implicated in controlling metastasis.55 They can stimulate naïve T cells and existing foreign antigens to helper T cells. Tumor-Associated Macrophages (TAMs) are brought to the tumor site by chemokines generated from stromal and cancer cells. Depending on their type of polarization, TAMs can perform one of two conflicting actions: M1-type TAMs release Interferon (IFN)-γ and IL-12 to stimulate the immune system and have pro-inflammatory and anti-tumoral effects. However, M2-type TAMs are pro-tumorigenic, produce IL-10, stimulate angiogenesis, and cause tumor cells to generate MMPs, which promote cancer growth through upsetting the BM and ECM.56,57
TAMs promote cancer cell survival and outgrowth at distant organs, activate the Epithelial-to-Mesenchymal Transition (EMT), localize invasion and intravasation into the bloodstream, transfer cancer cells via the circulation, extravasate and seed at the secondary location, and ultimately facilitate metastasis at multiple phases of the procedure.58 They accomplish this by releasing many chemokines, inflammatory chemicals, and growth components contributing to metastasis. TAMs assist in establishing an environment at the original tumor site conducive to tumor invasion.54 TMEM (Tumor Microenvironment of Metastasis) is introduced to designate the close integration of endothelial cells, perivascular TAMs, and cancer cells frequently found at intravasation locations. The likelihood of distant organ metastases is strongly correlated with increasing TMEM density in patient samples of breast cancer.54 Growth factors and cytokines, such as TGF-β, Wnt, and EGF, can trigger an organized transcriptional program that causes tumor cells to undergo Epithelial-Mesenchymal Transition (EMT), gaining mesenchymal properties and increasing their ability to invade and metastasis.58 There have also been reports of EMT caused by inflammation, and TAMs seem crucial to this transformation.
Regarding Hepatocellular Carcinoma (HCC), cancer cells release glypican, which attracts TAMs. They also emit TGF-β, Chemokine (C-C motif) Ligand 2 (CCL2), PDGF, VEGF, and M-CSF.54 TAMs release various cytokines, including IL-6 and TGF-β, into the tumor microenvironment, which can trigger EMT. Through TLR4/IL-10 signaling, M2-polarized TAMs expressing Toll-Like Receptor 4 (TLR4) facilitated EMT in pancreatic adenocarcinoma. PAMs specifically boosted the shape, proliferation, and migration of fibroblasts in pancreatic cancer cells by upregulating the mesenchymal markers vimentin and snail, inducing MMP-2 and MMP-9 proteolytic activity, and decreasing E-cadherin levels.54 Through the NF-κB/FAK pathway, TAMs with a CD68+HLA-DR+ surface marker phenotype can cause HCC cells to migrate. TAMs have the potential to enhance the invasiveness of cancer cells caused thru PRL-3 (phosphatase of the regenerating liver), a hallmark of liver metastasis in Colorectal Cancer (CRC).
Moreover, PRL-3 and Tumor Necrosis Factor-α (TNF-α) produced by CRC cells enhance the development of intermediate-conductance Ca2+-activated potassium (KCNN4) channels in TAMs; KCNN4 also causes TAMs to secrete IL-6 and IL-8, which increases the invasiveness of CRC cells. TAMs can also release the chemokine CCL18, which can increase angiogenesis and accelerate the growth of breast cancer tumors.59 Apart from their function in controlling the migration and invasion of primary tumor cells, TAMs also show a critical character in regulating the actions of cancer cells that have spread to secondary tissues.60
It has been suggested that the MAMs (Metastasis-Associated Macrophages) be used to characterize the function of macrophages that have colonized the metastatic location. Through the production of VEGF, MAMs play a crucial role in the extravasation of Circulating Tumor Cells (CTCs) and their effective expansion at the secondary location.54 Metastatic illness in breast cancer has been linked to CCL2 expression and macrophage infiltration of the tumor site.54 MAMs produced via Inflammatory Monocytes (IMs) are attracted to secondary locations in conjunction with monocytes that have the CCR2 receptor expressed on them. These cells are drawn to the metastatic location by the expression of CCL2 by both the tumor and the stroma. While CCR2 activation subsequent binding to CCL2 in MAMs induces the secretion of the chemokine ligand CCL3 thru macrophages at the metastatic location, allowing the retention of macrophages at the lung and increasing the number of lung metastatic foci, inhibition of CCR1, the CCL3 receptor, may have therapeutic implications in breast cancer lung metastasis.61 Furthermore, lung metastatic relapse has been linked to the expression of Vascular Cell Adhesion Molecule-1 (VCAM-1) in breast cancer cells.54 After breast cancer cells invade the lung’s leukocyte-rich milieu, VCAM-1 gives the cancer cells a survival improvement by means of attaching MAMs to them through counter-receptor α4 integrins.54
The role mesenchymal stem cells play in controlling metastasis
Multipotent stem cells known as Mesenchymal Stem Cells (MSCs) can be found in various adult tissues, including the bone marrow, adipose tissue, periosteum, lung, liver, muscle connective tissue, and spleen. They have a crucial role in developing and maintaining skeletal structures, including bone and cartilage.54 MSCs are seen in most tumors and significantly impact the TME’s growth and functionality. The tumor reprogrammes these Cancer-Associated MSCs (CA-MSC) to perform pro-tumorigenic activities, like boosting angiogenesis, metastasis, and EMT.
Crucially, MSCs promote metastases by secreting exosomes that influence the migration and proliferation of cancer cells.62,63 The ability to generate exosomes in large quantities is unique to them.54 MSC-derived exosomes are 60-200 nm-sized microvesicles with a phospholipid bilayer that contain mRNA, proteins, lipids, and miRNAs.54 They can be found in various bodily fluids and function in a paracrine manner.64 MSC-exosome therapy improved migratory capacity in a breast cancer model by upregulating β-catenin levels and activating the WNT way mark genes Dkk1and Axin2. Mesenchymal stem cells related to Gastric Cancer (GC-MSCs) release exosomes containing miRNAs that, when incorporated into gastric cancer cells, can stimulate the spread of gastric cancer. In particular, there was a considerable increase in miR-221 expression, linked with lymphatic metastasis, progressive tumor-node-metastasis stage, and more significant local invasion.54 In general, exosomes from MSCs triggered an EMT program and encouraged the migration and invasion of gastric cancer cells known as HGC-27.65 Mesenchymal Stem Cells generated from Bone Marrow (BM-MSCs) can preferentially transport cytokines, including chemotactic proteins 40 SDF-1, 41, 42, MCP-1, MCP-2, MCP-3, and IGF-1, which can aid in the promotion of multiple myeloma cell migration.64
In contrast to results, MSCs were likewise discovered to inhibit metastatic tumor formation by excreting exosomes containing distinct strands of miRNAs. MSCs can support cancer cell dormancy and allow for an extended period of cycling dormancy, keeping cancer cells in the G0/G1 phase of the cell cycle by interacting by dispersed breast cancer cells in the bone marrow throughout the initial dissemination phases.66,67 Exosomes produced by MSCs that express more miR-23b and less MARCKS reduce the cell cycle and encourage the dormancy of breast cancer cells that have spread.54,68
Therapeutics approach to cancer metastasis for individuals who are considered curable at diagnosis, surgery is still the principal cancer therapy option available today. Current surgical techniques can successfully remove most tumors. Still, cancer cells that were removed throughout surgery or that had previously moved outside of the original tumor locations are significant sources of metastasis. Later, the migrating cells compromise the function at the afresh metastasized organ locations. Cancer-related mortality eventually results from functional impairment at metastatic locations.69
Significant progress has been achieved in the areas of cancer screening, early detection, and the creation of innovative chemotherapy and radiation treatment plans. On the other hand, not much has changed in terms of preventing cancer or stopping primary tumors from spreading. Consequently, a multifaceted strategy is required to stop the primary tumor from getting propagated. Inhibiting metastasis and halting tumor development are two crucial characteristics of novel anticancer medications. Chemotherapy medications were first created to treat primary malignancies. We have progressed from monotherapy to regimens of combination treatment. The chemotherapy toolbox has expanded to include several novel drugs throughout the last ten years. These novel medications focus on particular cell signaling cascades. Kinase inhibitors and monoclonal antibodies are two of the emerging targeted agents. These novel medications work best when combined with existing antimetabolite medications, such as 5-fluorouracil, rather than by themselves. This emphasizes the requirement for additional medications that could target primary tumors.69
Human clinical trials are only accessed by 5% of small molecules (investigational medicines) possessing therapeutic characteristics. Several experimental medications are removed from clinical trials because of their toxicity or ineffectiveness. The trial medications are rarely taken on their own. They are instead added to already-approved medications. Clinical trials also aim to lower the primary tumor burden. However, these experimental medications may prevent metastases. As a result, we need to redouble our efforts to target goals other than lowering the primary tumor burden. For instance, EMT is damaging to cancer patients since it is a defining characteristic of cancer cells that have metastasized, despite its importance in development and wound repair. Therefore, a critical field of research toward creating treatments is to focus on ways to stop EMT in cancer cells. One helpful strategy might be to create a list of EMT-affected markers. A panel like this might be used as a standard by tiny molecular screens to identify compounds with anti-EMT characteristics. These small compounds are typically not cytotoxic, but they may interact with important EMT regulators, among other things, to produce antimetastatic effects. Cancer patients may benefit from combining the minor compounds found through these screens with standard-of-care medications.
Many currently used anticancer medicines were first created from natural materials. The three main places where novel drugs are discovered are plants, fungi, and marine creatures. Ranges of 60-85% of currently used chemotherapeutic drugs are derivatives of natural products.69 However, not every bioactive compound extracted from a natural product finds its way into a clinical setting since some of these compounds may be hazardous or fail to stop the proliferation of cancer cells. The small compounds that were separated, however, might block particular signaling pathways that support the growth of tumors. The current focus of anticancer medication discovery is toxicity to tumor cells. These methods will not catch specific inhibitory actions of the test substances. As a result, we also need to screen small compounds for antimetastatic qualities thoroughly. The resulting molecules can be coupled with other cytotoxic medications to prevent metastasis (Figure 3).
Natural product drug development faces two significant obstacles. The technical one comes first. Government rules about biodiversity are the second and just as substantial. Several compounds working together in unrefined preparations are a significant technical challenge. In vitro synthesis of natural compounds has proven challenging, even in cases where a single active molecule has been discovered. Difficulties with Intellectual Property (IP) and the environment impact buying natural products domestically and abroad. They become vital when considering the Biodiversity Convention’s rules and regulations in a larger context. The discovery and development of novel treatment approaches to address cancer metastasis hold considerable promise, notwithstanding these challenges. Much remains about this field, although many substances have shown anticancer potential (Table 3).
| Priority targets for various cancer hallmarks such as tissue invasion and metastasis | Upregulation of E-cadherin | Inhibition of PI3K/AKT signalling | Inhibition of FAK signalling | Reference |
|---|---|---|---|---|
| Genomic Instability | (+) | (+) | (0) | 70 |
| Sustained Proliferative Signalling | (+/-) | (+) | (+) | 70 |
| Tumor promoting inflammation | (+/-) | (+) | (+) | 70 |
| Evasion of anti-growth signalling | (+/-) | (+) | (+) | 70 |
| Resistance to apoptosis | (+) | (+) | (+) | 70 |
| Replicative immortality | (0) | (+/-) | (+) | 70 |
| Angiogenesis | (-) | (+) | (+) | 70 |
| Immune system evasion | (0) | (+/-) | (0) | 70 |
Important targets found in metastasis and tissue invasion were also investigated in the other cancer characteristic areas.
Future perspective
The multifaceted process of cancer spreading depends on extrinsic mechanisms that create the immunosuppressive stromal milieu and intrinsic networks that support plasticity and stress responses. Although treating primary tumors with driver-centric techniques results in less complex treatment requirements, the possibility of tumor type-agnostic treatments is vital due to the convergent evolution of different tumor types during the metastatic process. Some common motifs show that cellular plasticity and stress-relieving pathways are essential for metastatic competency despite the complexity and diversity of the paths and processes causing metastasis. Notably, a few potential targets common to all cancer types have been confirmed to represent important regulatory nodes. Consequently, a biomarker-driven method that validates single agent or combinatorial approaches based on the molecular makeup of individual tumors should be used to treat metastatic malignancies. Recent research has demonstrated that a critical strategy to increase overall survival would involve tumor-intrinsic targeting and the simultaneous reduction of immune suppressive proteins. Preventing metastasis-associated resistance and enhancing long-term responses will also require combinational medicines with different modes of action.71
Finding treatments that enforce metastatic retreat has received relatively little attention in the preclinical context. Even though reaching this threshold in preclinical models is the hardest, these proof-of-concept measurements should be vital to the efficacy data to guide early drug development. Furthermore, regulatory flexibility will be necessary to progress these emerging kinds of treatments in a way that maximizes resource use. Predictive pharmacodynamic markers and 1-year PFS/OS measurements may be used for high-need patients as secondary outcome measures. This is especially crucial since high-risk patients frequently switch between different trial regimens, making overall survival challenging.71
The significance of lifestyle and diagnostic factors in lowering cancer mortality must also be prioritized in searching for novel treatments. The greatest threat to our recent gains in cancer is the upsurge in obesity rates, becoming the most significant etiologic cause of cancer and metastasis initiation.72,73 Meanwhile, throughout much of the world, other avoidable dangers continue to be a major cause of cancer. A significant increase in survival will come from better diagnostic and early detection efforts for malignancies whose metastases develop slowly. To further lower cancer mortality, preventive, early diagnosis, and treatment strategies must all be thoroughly optimized.72,73
CONCLUSION
This article discusses about, the complex process of tumor invasion and metastasis, which is a multidimensional phenomenon involving many cellular and molecular processes. The process known as the epithelial-mesenchymal transition (EMT) is essential for giving cancer cells the ability to migrate and invade, which frees them from the boundaries of the original tumor site. Adhesion molecules in healthy tissues control cell adhesion, which in turn controls how cancer cells interact with their surroundings and affects the start and course of metastasis. Cancer cells can spread to distant locations more easily when new blood and lymphatic vessels are formed as a result of angiogenesis and lymphangiogenesis. Whether by hematogenous or lymphatic pathways, the extravasation of tumor cells represents a crucial stage in the metastatic cascade. The LKB1/AMPK signaling pathway is tangled in complicated regulatory mechanisms that highlight the intricacy of tumor invasion and migration, which are governed by the convergence of several signaling pathways. The behavior of cancer cells is significantly shaped by the tumor microenvironment, which also affects the cells capacity to invade and spread. Mesenchymal stem cells play a vital role in the dynamic interactions that occur within the tumor microenvironment and are actively involved in the regulation of metastasis. Targeted treatment interventions become possible when the molecular details and different cell types involved in this process are understood. The treatment of cancer metastasis is changing, with an emphasis on modifying the tumor microenvironment, blocking important signaling pathways, and concentrating on particular stages of the metastatic cascade. Future developments in the understanding of metastasis may entail combination and customized treatments that take into account the particulars of each patient’s tumor. To put it simply, understanding the molecular and cellular mechanisms driving tumor invasion and metastasis lays the groundwork for creating cutting-edge and successful treatment approaches. As we learn more about the intricacies of cancer biology, there is hope for bettering patient outcomes and reducing the worldwide cancer burden through the development of novel therapies and preventive measures against metastasis.
Cite this article:
Asiri A, Ali AA, Althobiti MM, Abu-Alghayth MH. Tumor Invasion and Metastasis: A Current and Emerging Therapeutic Approaches. Int. J. Pharm. Investigation. 2024;14(4):983-97.
ACKNOWLEDGEMENT
The authors are thankful to the Deanship of Graduate Studies and Scientific Research at the University of Bisha for supporting this work through the Fast-Track Research Support Program.
ABBREVIATIONS
| TME | Tumor microenvironment |
|---|---|
| ECM | Extracellular matrix |
| EMT | Epithelial-mesenchymal transition |
| CTCs | Circulating tumor cells |
| LN | Lymph nodes |
| EGF | Epidermal growth factor |
| HBEGF | Heparin-binding EGF-like growth factor |
| COX2 | Cyclooxygenase 2 |
| APP | Amyloid precursor protein |
| DR6 | Death receptor 6 |
| PTEN | Phosphatase and tensin homolog removed in chromosome ten |
| LPA | Lysophosphatidic acid |
| CCL-3 | Chemokine ligand 3 |
| LITAF | Lipopolysaccharide-induced TNF-α factor |
| IGF-1 | Insulin-like growth factor 1 |
| EVs | Extracellular vesicles |
| TAMs | Tumor-associated macrophages |
| HSC | Hematopoietic stem cells |
| MSCs | Mesenchymal stem cells |
References
- Massagué J, Obenauf AC. Metastatic colonization by circulating tumour cells. Nature. 2016;529(7586):298-306. [PubMed] | [CrossRef] | [Google Scholar]

- Ye X, Weinberg RA. Epithelial-mesenchymal plasticity: a central regulator of cancer progression. Trends Cell Biol. 2015;25(11):675-86. [PubMed] | [CrossRef] | [Google Scholar]
- Fares J, Fares MY, Khachfe HH, Salhab HA, Fares Y. Molecular principles of metastasis: a hallmark of cancer revisited. Signal Transduct Target Ther. 2020;5(1):28 [PubMed] | [CrossRef] | [Google Scholar]
- Maitra A. Molecular envoys pave the way for pancreatic cancer to invade the liver. Nature. 2019;567(7747):181-2. [PubMed] | [CrossRef] | [Google Scholar]
- Fouad YA, Aanei C. Revisiting the hallmarks of cancer. Am J Cancer Res. 2017;7(5):1016-36. [PubMed] | [Google Scholar]
- Knott SR, Wagenblast E, Khan S, Kim SY, Soto M, Wagner M, et al. Asparagine bioavailability governs metastasis in a model of breast cancer. Nature. 2018;554(7692):378-81. [PubMed] | [CrossRef] | [Google Scholar]
- Nieto MA, Huang RY, Jackson RA, Thiery JP. EMT: 2016. Cell. 2016;166(1):21-45. [PubMed] | [CrossRef] | [Google Scholar]
- Pastushenko I, Brisebarre A, Sifrim A, Fioramonti M, Revenco T, Boumahdi S, et al. Identification of the tumour transition states occurring during EMT. Nature. 2018;556(7702):463-8. [PubMed] | [CrossRef] | [Google Scholar]
- Erdogan B, Ao M, White LM, Means AL, Brewer BM, Yang L, et al. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J Cell Biol. 2017;216(11):3799-816. [PubMed] | [CrossRef] | [Google Scholar]
- Rankin EB, Giaccia AJ. Hypoxic control of metastasis. Science. 2016;352(6282):175-80. [PubMed] | [CrossRef] | [Google Scholar]
- Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST, et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 2015;527(7579):472-6. [PubMed] | [CrossRef] | [Google Scholar]
- Zheng XF, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527(7579):525-30. [PubMed] | [CrossRef] | [Google Scholar]
- Diepenbruck M, Christofori G. Epithelial-mesenchymal transition (EMT) and metastasis: yes, no, maybe?. Curr Opin Cell Biol. 2016;43:7-13. [PubMed] | [CrossRef] | [Google Scholar]
- Esposito M, Mondal N, Greco TM, Wei Y, Spadazzi C, Lin SC, et al. Bone vascular niche E-selectin induces mesenchymal-epithelial transition and Wnt activation in cancer cells to promote bone metastasis. Nat Cell Biol. 2019;21(5):627-39. [PubMed] | [CrossRef] | [Google Scholar]
- Shibue T, Weinberg RA. EMT, CSCS, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14(10):611-29. [PubMed] | [CrossRef] | [Google Scholar]
- Lu J, Kornmann M, Traub B. Role of epithelial to mesenchymal transition in colorectal cancer. Int J Mol Sci. 2023;24(19):14815 [PubMed] | [CrossRef] | [Google Scholar]
- Ribatti D, Tamma R, Annese T. Epithelial-mesenchymal transition in cancer: A historical overview. Transl Oncol. 2020;13(6):100773 [PubMed] | [CrossRef] | [Google Scholar]
- Zadeh MM, Motamed N, Ranji N, Majidi M, Falahi F. Silibinin-induced apoptosis and downregulation of microRNA-21 and microRNA-155 in MCF-7 human breast cancer cells. J Breast Cancer. 2016;19(1):45-52. [PubMed] | [CrossRef] | [Google Scholar]
- Chen Z, Yuan YC, Wang Y, Liu Z, Chan HJ, Chen S, et al. Down-regulation of programmed cell death 4 (PDCD4) is associated with aromatase inhibitor resistance and a poor prognosis in estrogen receptor-positive breast cancer. Breast Cancer Res Treat. 2015;152(1):29-39. [PubMed] | [CrossRef] | [Google Scholar]
- Bahena-Ocampo I, Espinosa M, Ceballos-Cancino G, Lizarraga F, Campos-Arroyo D, Schwarz A, et al. miR-10b expression in breast cancer stem cells supports self-renewal through negative PTEN regulation and sustained AKT activation. EMBO Rep. 2016;17(5):648-58. [PubMed] | [CrossRef] | [Google Scholar]
- Liang AL, Zhang TT, Zhou N, Wu CY, Lin MH, Liu YJ, et al. miRNA-10b sponge: an anti-breast cancer study
. Oncol Rep. 2016;35(4):1950-8. [PubMed] | [CrossRef] | [Google Scholar] - Petrović N. miR-21 might be involved in breast cancer promotion and invasion rather than in initial events of breast cancer development. Mol Diagn Ther. 2016;20(2):97-110. [PubMed] | [CrossRef] | [Google Scholar]
- Wu ZH, Tao ZH, Zhang J, Li T, Ni C, Xie J, et al. MiRNA-21 induces epithelial to mesenchymal transition and gemcitabine resistance via the PTEN/AKT pathway in breast cancer. Tumour Biol. 2016;37(6):7245-54. [PubMed] | [CrossRef] | [Google Scholar]
- De Mattos-Arruda L, Bottai G, Nuciforo PG, Di Tommaso LD, Giovannetti E, Peg V, et al. MicroRNA-21 links epithelial-to-mesenchymal transition and inflammatory signals to confer resistance to neoadjuvant trastuzumab and chemotherapy in HER2-positive breast cancer patients. Oncotarget. 2015;6(35):37269-80. [PubMed] | [CrossRef] | [Google Scholar]
- Yu DD, Lv MM, Chen WX, Zhong SL, Zhang XH, Chen L, et al. Role of miR-155 in drug resistance of breast cancer. Tumour Biol. 2015;36(3):1395-401. [PubMed] | [CrossRef] | [Google Scholar]
- Zhao M, Ang L, Huang J, Wang J. MicroRNAs regulate the epithelial-mesenchymal transition and influence breast cancer invasion and metastasis. Tumour Biol. 2017;39(2):1010428317691682 [PubMed] | [CrossRef] | [Google Scholar]
- Kwak SY, Yoo JO, An HJ, Bae IH, Park MJ, Kim J, et al. miR-5003-3p promotes epithelial-mesenchymal transition in breast cancer cells through Snail stabilization and direct targeting of E-cadherin. J Mol Cell Biol. 2016;8(5):372-83. [PubMed] | [CrossRef] | [Google Scholar]
- Yoo JO, Kwak SY, An HJ, Bae IH, Park MJ, Han YH, et al. miR-181b-3p promotes epithelial-mesenchymal transition in breast cancer cells through Snail stabilization by directly targeting YWHAG. Biochim Biophys Acta. 2016;1863(7 Pt A):1601-11. [PubMed] | [CrossRef] | [Google Scholar]
- Rasheed SA, Teo CR, Beillard EJ, Voorhoeve PM, Zhou W, Ghosh S, et al. MicroRNA-31 controls G protein alpha-13 (GNA13) expression and cell invasion in breast cancer cells. Mol Cancer. 2015;14:67 [PubMed] | [CrossRef] | [Google Scholar]
- Lu Z, Jiao D, Qiao J, Yang S, Yan M, Cui S, et al. Restin suppressed epithelial-mesenchymal transition and tumor metastasis in breast cancer cells through upregulating mir-200a/b expression via association with p73. Mol Cancer. 2015;14:102 [PubMed] | [CrossRef] | [Google Scholar]
- Kong X, Ding X, Li X, Gao S, Yang Q. 53BP1 suppresses epithelial-mesenchymal transition by downregulating ZEB1 through microRNA-200b/429 in breast cancer. Cancer Sci. 2015;106(8):982-9. [PubMed] | [CrossRef] | [Google Scholar]
- Liu J, Zhou Y, Shi Z, Hu Y, Meng T, Zhang X, et al. microRNA-497 modulates breast cancer cell proliferation, invasion, and survival by targeting SMAD7. DNA Cell Biol. 2016;35(9):521-9. [PubMed] | [CrossRef] | [Google Scholar]
- Hong L, Pan F, Jiang H, Zhang L, Liu Y, Cai C, et al. miR-125b inhibited epithelial-mesenchymal transition of triple-negative breast cancer by targeting MAP2K7. Onco Targets Ther. 2016;9:2639-48. [PubMed] | [CrossRef] | [Google Scholar]
- Li G, Yao L, Zhang J, Li X, Dang S, Zeng K, et al. Tumor-suppressive microRNA-34a inhibits breast cancer cell migration and invasion via targeting oncogenic TPD52. Tumour Biol. 2016;37(6):7481-91. [PubMed] | [CrossRef] | [Google Scholar]
- Yin K, Yin W, Wang Y, Zhou L, Liu Y, Yang G, et al. MiR-206 suppresses epithelial mesenchymal transition by targeting TGF-β signaling in estrogen receptor positive breast cancer cells. Oncotarget. 2016;7(17):24537-48. [PubMed] | [CrossRef] | [Google Scholar]
- Zhang J, Liu D, Feng Z, Mao J, Zhang C, Lu Y, et al. RETRACTED: MicroRNA-138 modulates metastasis and EMT in breast cancer cells by targeting vimentin. Biomed Pharmacother. 2016;77:135-41. [PubMed] | [CrossRef] | [Google Scholar]
- Singh R, Yadav V, Kumar S, Saini N. MicroRNA-195 inhibits proliferation, invasion and metastasis in breast cancer cells by targeting FASN, HMGCR, ACACA and CYP27B1. Sci Rep. 2015;5:17454 [PubMed] | [CrossRef] | [Google Scholar]
- Mui KL, Chen CS, Assoian RK. The mechanical regulation of integrin-cadherin crosstalk organizes cells, signaling and forces. J Cell Sci. 2016;129(6):1093-100. [PubMed] | [CrossRef] | [Google Scholar]
- Janiszewska M, Primi MC, Izard T. Cell adhesion in cancer: beyond the migration of single cells. J Biol Chem. 2020;295(8):2495-505. [PubMed] | [CrossRef] | [Google Scholar]
- Shashikanth N, Petrova YI, Park S, Chekan J, Maiden S, Spano M, et al. Allosteric regulation of E-cadherin adhesion. J Biol Chem. 2015;290(35):21749-61. [PubMed] | [CrossRef] | [Google Scholar]
- Bachmann M, Kukkurainen S, Hytönen VP, Wehrle-Haller B. Cell adhesion by integrins. Physiol Rev. 2019;99(4):1655-99. [PubMed] | [CrossRef] | [Google Scholar]
- Aksorn N, Chanvorachote P. Integrin as a molecular target for anti-cancer approaches in lung cancer. Anticancer Res. 2019;39(2):541-8. [PubMed] | [CrossRef] | [Google Scholar]
- Sun Z, Tseng HY, Tan S, Senger F, Kurzawa L, Dedden D, et al. Kank2 activates talin, reduces force transduction across integrins and induces central adhesion formation. Nat Cell Biol. 2016;18(9):941-53. [PubMed] | [CrossRef] | [Google Scholar]
- Bielenberg DR, Zetter BR. The contribution of angiogenesis to the process of metastasis. Cancer J. 2015;21(4):267-73. [PubMed] | [CrossRef] | [Google Scholar]
- Guillerey C, Smyth MJ. NK cells and cancer immunoediting. Curr Top Microbiol Immunol. 2016;395:115-45. [PubMed] | [CrossRef] | [Google Scholar]
- Deryugina EI, Quigley JP. Tumor angiogenesis: MMP-mediated induction of intravasation- and metastasis-sustaining neovasculature. Matrix Biol. 2015:44-46. [PubMed] | [CrossRef] | [Google Scholar]
- Beije N, Jager A, Sleijfer S. Circulating tumor cell enumeration by the CellSearch system: the clinician’s guide to breast cancer treatment?. Cancer Treat Rev. 2015;41(2):144-50. [PubMed] | [CrossRef] | [Google Scholar]
- Strilic B, Offermanns S. Intravascular survival and extravasation of tumor cells. Cancer Cell. 2017;32(3):282-93. [PubMed] | [CrossRef] | [Google Scholar]
- Strilic B, Yang L, Albarrán-Juárez J, Wachsmuth L, Han K, Müller UC, et al. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature. 2016;536(7615):215-8. [PubMed] | [CrossRef] | [Google Scholar]
- Tichet M, Prod’Homme V, Fenouille N, Ambrosetti D, Mallavialle A, Cerezo M, et al. Tumour-derived SPARC drives vascular permeability and extravasation through endothelial VCAM1 signalling to promote metastasis. Nat Commun. 2015;6:6993 [PubMed] | [CrossRef] | [Google Scholar]
- Pandey P, Sliker B, Peters HL, Tuli A, Herskovitz J, Smits K, et al. Amyloid precursor protein and amyloid precursor-like protein 2 in cancer. Oncotarget. 2016;7(15):19430-44. [PubMed] | [CrossRef] | [Google Scholar]
- Chiang SP, Cabrera RM, Segall JE. Tumor cell intravasation. Am J Physiol Cell Physiol. 2016;311(1):C1-C14. [PubMed] | [CrossRef] | [Google Scholar]
- Kitamura T, Qian BZ, Pollard JW. Immune cell promotion of metastasis. Nat Rev Immunol. 2015;15(2):73-86. [PubMed] | [CrossRef] | [Google Scholar]
- Christiana M, Neophytou MP, Triantafyllos S, Panagiotis P. The role of tumor microenvironment in cancer metastasis: molecular mechanisms and therapeutic opportunities. Cancers. 2021;13(9):2053 [PubMed] | [CrossRef] | [Google Scholar]
- Neophytou CM, Pierides C, Christodoulou MI, Costeas P, Kyriakou TC, Papageorgis P, et al. The role of tumor-associated my-eloid cells in modulating cancer therapy. Front Oncol. 2020;10:899 [PubMed] | [CrossRef] | [Google Scholar]
- Whipple CA. Tumor talk: understanding the conversation between the tumor and its microenvironment. Cancer Cell Microenviron. 2015;2(2) [PubMed] | [CrossRef] | [Google Scholar]
- Singh S, Mehta N, Lilan J, Budhthoki MB, Chao F, Yong L, et al. Initiative action of tumor-associated macrophage during tumor metastasis. Biochim Open. 2017;4:8-18. [PubMed] | [CrossRef] | [Google Scholar]
- Sanchez LR, Borriello L, Entenberg D, Condeelis JS, Oktay MH, Karagiannis GS, et al. The emerging roles of macrophages in cancer metastasis and response to chemotherapy. J Leukoc Biol. 2019;106(2):259-74. [PubMed] | [CrossRef] | [Google Scholar]
- Papageorgis P. TGFbeta signaling in tumor initiation, epithelial-to-mesenchymal transition, and metastasis. J Oncol. 2015;2015:587193 [PubMed] | [CrossRef] | [Google Scholar]
- Lin L, Chen YS, Yao YD, Chen JQ, Chen JN, Huang SY, et al. CCL18 from tumor-associated macrophages promotes angiogenesis in breast cancer. Oncotarget. 2015;6(33):34758-73. [PubMed] | [CrossRef] | [Google Scholar]
- Lin Y, Xu J, Lan H. Tumor-associated macrophages in tumor metastasis: biological roles and clinical therapeutic applica-tions. J Hematol Oncol. 2019;12(1):76 [PubMed] | [CrossRef] | [Google Scholar]
- Kitamura T, Qian BZ, Soong D, Cassetta L, Noy R, Sugano G, et al. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J Exp Med. 2015;212(7):1043-59. [PubMed] | [CrossRef] | [Google Scholar]
- Zhou J, Tan X, Tan Y, Li Q, Ma J, Wang G, et al. Mesenchymal stem cell derived exosomes in cancer progression, metastasis and drug delivery: A comprehensive review. J Cancer. 2018;9(17):3129-37. [PubMed] | [CrossRef] | [Google Scholar]
- Atiya H, Frisbie L, Pressimone C, Coffman L. Mesenchymal stem cells in the tumor microenvironment. Adv Exp Med Biol. 2020;1234:31-42. [PubMed] | [CrossRef] | [Google Scholar]
- Li N, Huang D, Lu N, Luo L. Role of the LKB1/AMPK pathway in tumor invasion and metastasis of cancer cells (Review) [review]. Oncol Rep. 2015;34(6):2821-6. [PubMed] | [CrossRef] | [Google Scholar]
- Gu H, Ji R, Zhang X, Wang M, Zhu W, Qian H, et al. Exosomes derived from human mesenchymal stem cells promote gastric cancer cell growth and migration via the activation of the Akt pathway. Mol Med Rep. 2016;14(4):3452-8. [PubMed] | [CrossRef] | [Google Scholar]
- Casson J, Davies OG, Smith CA, Dalby MJ, Berry CC. Mesenchymal stem cell-derived extracellular vesicles may promote breast cancer cell dormancy. J Tissue Eng. 2018;9:2041731418810093 [PubMed] | [CrossRef] | [Google Scholar]
- Neophytou CM, Kyriakou TC, Papageorgis P. Mechanisms of metastatic tumor dormancy and implications for cancer therapy. Int J Mol Sci. 2019;20(24):6158 [PubMed] | [CrossRef] | [Google Scholar]
- Jiang WG, Sanders AJ, Katoh M, Ungefroren H, Gieseler F, Prince M, et al. Tissue invasion and metastasis: molecular, biological and clinical perspectives. Semin Cancer Biol. 2015;35(Suppl) [PubMed] | [CrossRef] | [Google Scholar]
- Bliss SA, Sinha G, Sandiford OA, Williams LM, Engelberth DJ, Guiro K, et al. Mesenchymal stem cell-derived exosomes stimulate cycling quiescence and early breast cancer dormancy in bone marrow. Cancer Res. 2016;76(19):5832-44. [PubMed] | [CrossRef] | [Google Scholar]
- Jiang WG, Sanders AJ, Katoh M, Ungefroren H, Gieseler F, Prince M, et al. Tissue invasion and metastasis: molecular, biological and clinical perspectives. Semin Cancer Biol. 2015;35(Suppl) [PubMed] | [CrossRef] | [Google Scholar]
- Esposito M, Ganesan S, Kang Y. Emerging strategies for treating metastasis. Nat Cancer. 2021;2(3):258-70. [PubMed] | [CrossRef] | [Google Scholar]
- Quail DF, Olson OC, Bhardwaj P, Walsh LA, Akkari L, Quick ML, et al. Obesity alters the lung myeloid cell landscape to enhance breast cancer metastasis through IL5 and GM-CSF. Nat Cell Biol. 2017;19(8):974-87. [PubMed] | [CrossRef] | [Google Scholar]