ABSTRACT
The present study describes the process of regulatory submission and generic drug registration in Europe, the UK, Australia, and New Zealand. The information and data for the compilation of the present review has been obtained from the relevant journals and from the official websites of respective drug regulatory authorities of Europe, the UK, Australia, and New Zealand. The generic drug approval process differs from one country to another. Europe follows multiple registration processes and the applicant decides the pathway based on the product category while the other three countries stick to a single registration process. All four countries follow the electronic CTD format for submitting the regulatory documents which allows the evaluation process more convenient and easier. The British Pharmacopoeia is a single reference guide for the quality control of medicines in all four countries under this study. From this study, it was concluded that in Europe and UK it takes about 12 months for dossier review for the approval of the generic drug. TGA of Australia takes approximately 11 months of timeline for generic drug approval whereas; MEDSAFE of New Zealand takes just 200 calendar days for generic drug registration which is quicker as compared to the other three countries.
INTRODUCTION
A drug is said to be “generic” if it has been developed to be exactly like a branded drug with regards to the dosage form, strength, and route of administration and intended use that has previously been marketed. Every country has its own rules and guidelines for generic drug registration.1
In United Kingdom (UK), the generic drugs are regulated and assessed by the drug regulatory authority named Medicines and Healthcare Products Regulatory Agency (MHRA). Generic drugs prescribing is seen as desirable in UK. The generic drug substitution is a standard practice by pharmacists in UK hospitals. In United Kingdom, most drugs are prescribed by their generic names in hospitals, and hence it is becoming more common in general practice. In comparison to a wide range of developed countries, both inside and outside Europe, the UK has some of the lowest drug prices for generics.2
European Medicines Agency (EMA) is the regulatory authority of European Union (EU). There are different regulatory approaches in EU for getting marketing authorization (MA) for generic drugs which are Centralized Procedure (CP), Decentralized Procedure (DCP), Mutual Recognition Procedure (MRP) and National Procedure (NP). The majority of MA submissions for generics in Europe happen through DCP and MRP approaches. Approval time for marketing authorisation application (MAA) is 12 months. Application fees ranges from 10-20 lakhs as fixed by EMA.3
Therapeutic Goods Administration (TGA) is the drug regulatory authority to regulate the generic drug products in Australia. Before a generic drug can be registered in Australia, it has to be included in the Pharmaceutical Benefits Scheme (PBS). Currently there has been inclusion of large number of generic drugs in the PBS portal. Generally, in Australia number of generic drugs are marketed by the brand names. TGA is a global standard complies with European and US drug regulations. Australia is a tough investment opportunity for multi-national generic companies that are designed for big volume markets due to the market’s small size and high cost of registration. TGA takes approximately 11 months for generic drug approval in Australia.4
In New Zealand all the medicinal products including generic drugs are regulated by Medicines and Medical Devices Safety Authority (MEDSAFE) which is a part of Ministry of Health of New Zealand. Very few bioequivalence studies are undertaken in New Zealand for generics along with dissolution studies. New Zealand’s trending system and use of preferred medicines resulted in low generic prices well before patent expiry. MEDSAFE takes only 200 calendar days for the generic drug approval.5
All the four countries use electronic Common Technical Document (eCTD) format for submitting the documents which allows the evaluation process more convenient and easier. General dossier requirements for generic drug registration in EU, UK, Australia and New Zealand are given below.
Generic drug registration dossier requirements
Module 1-Administrative information
Pre-Submission Planning Form (PPF)- Applicable only for Australia.
Signature of different types of Letters of Authorization (LOA).
Marketing Authorization Application (MAH).
Pharmacovigilance information document.
Information of designated person/site wishes to commercialize the product on the market.
Establishment proof certificate/letter.
Cover letter in local language (with signature of applicant).
Applicant form signed by the MAH in the Reference Member State (RMS).
Originally signed confirmation of identical dossier.
Documents/Statements to be provided in original or as legalized copies.
Copy of Trade mark.
Samples (finished product and API) to be submitted for analysis (Control).
Declaration of conformity of national translations of the SmPC, PI and labeling.
Bioavailability/Bioequivalence (BA/BE) data.
Certificate of Suitability (CEP)- Applicable only in Europe.
Module 2-Common Technical Document Summaries
Quality Overall Summary.
Non-clinical overview.
Clinical Summary.
Module 3-(Quality)
Quality information.
Module 4-Non-Clinical Study Reports
Module 5-Clinical Study Reports6-11
The eCTD triangle for the generic drug dossier submission is shown in Figure 1.
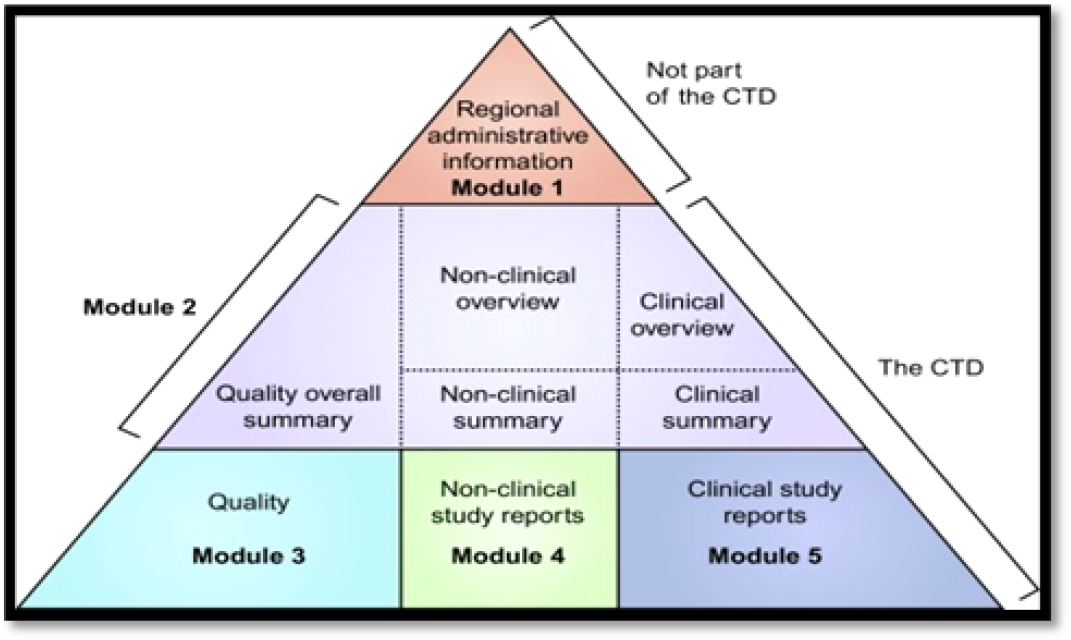
Figure 1:
The eCTD triangle with modules.
Generic drug approval process in European Union (EU)
In Europe, generic drugs are approved through 4 marketing authorization procedures. They are,
Centralized Procedure (CP).
National Procedure (NP).
Mutual Recognition Procedure (MRP).
Decentralized Procedure (DP).
Centralized Procedure
The process of CP approval has been described in EU regulation no. 724/2004 under the EU directive 2004/27/EC. At first, applicant has to send the proposal to the EMA, and then later EU Commission issues a European marketing authorization within 7 months (210 days) of the receipt of application. Initially the applicant has to submit the Marketing Authorization Application (MAA) to EMA. The received MAA dossier will be reviewed by the Committee for Human Medicinal Product (CHMP) and provides comments after 115th day of proposal submission. CHMP forwards the list of questions to the applicant on 120th day followed by ‘stop clock’. During this period, the evaluation of dossier is officially stopped, while the applicant prepares responses to quires received from the CHMP. The time resumes when the applicant has to send the responses to questions raised by CHMP. Joint assessment report will be sent by (co)-Rapporteur to the CHMP on 150th day of initial application submission. CHMP comments to this report on 170th of initial application submission followed by CHMP decision on need of oral explanation by the applicant. On 181st day of initial application submission, oral explanation will be given by applicant. On 185th day of initial application submission, the applicant presents the final draft to (co)-Rapporteur, EMA and CHMP. Once the final draft has been received by the authorities, CHMP decides its opinion regarding approval or rejection of MAA which takes nearly 210 days of timeline.12 The Centralized procedure for generic drug approval in EU has been depicted in Figure 2.
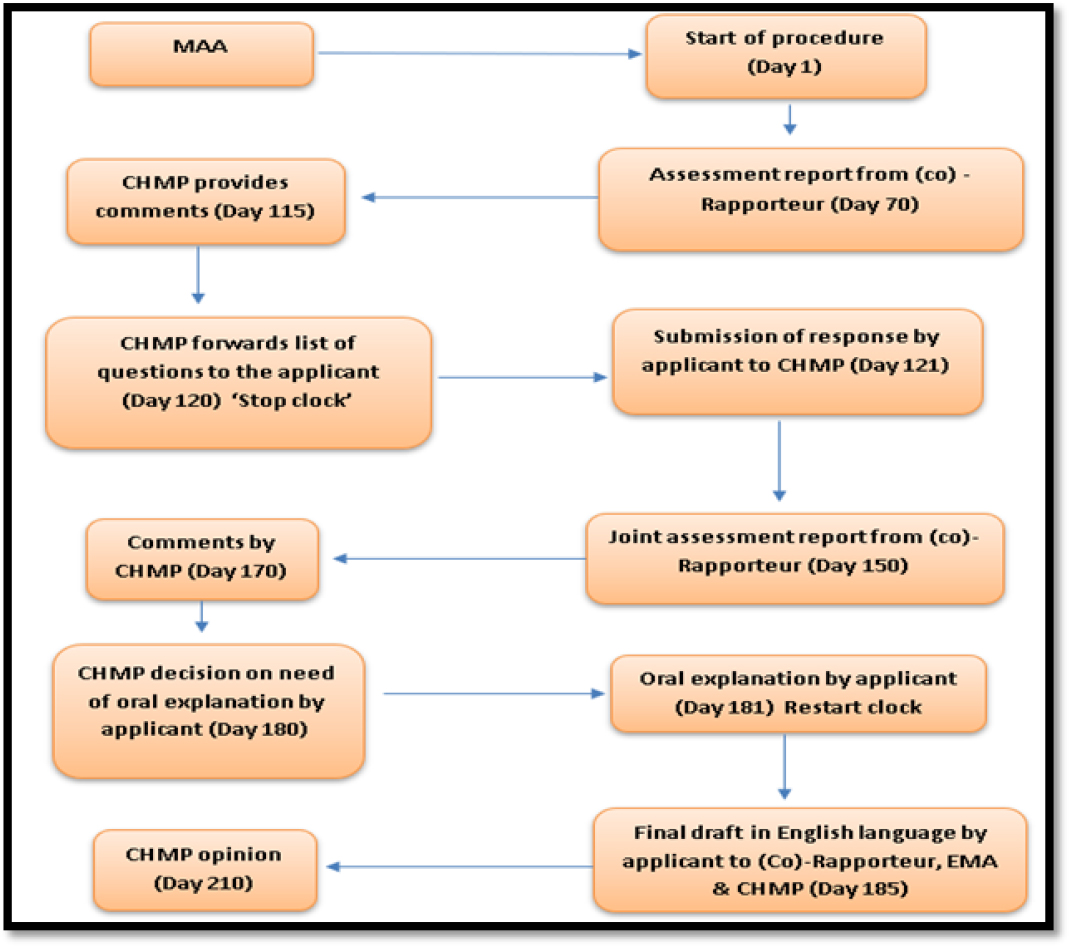
Figure 2:
Centralized procedure in EU for Generic drug approval.
National Procedure
This procedure is required whenever a company wants to commercialize a product in only one member state of Europe, which is followed prior to Mutual Reorganization Procedure (MRP). Applicant submits the application to competent authority in its native national language. Competent authority of Concerned Member State (CMS) will adopt a decision after thorough evaluation of application and produce an assessment report. In case, if the submitted application is incomplete, the authority will notify the applicant after 30 days of initial submission. If the submitted application fulfils all the requirements, competent authority will approve the application where applicant can wish to market the generic drug in single European country.12
The National Procedure (NP) for generic drug approval process in EU has been depicted in Figure 3.
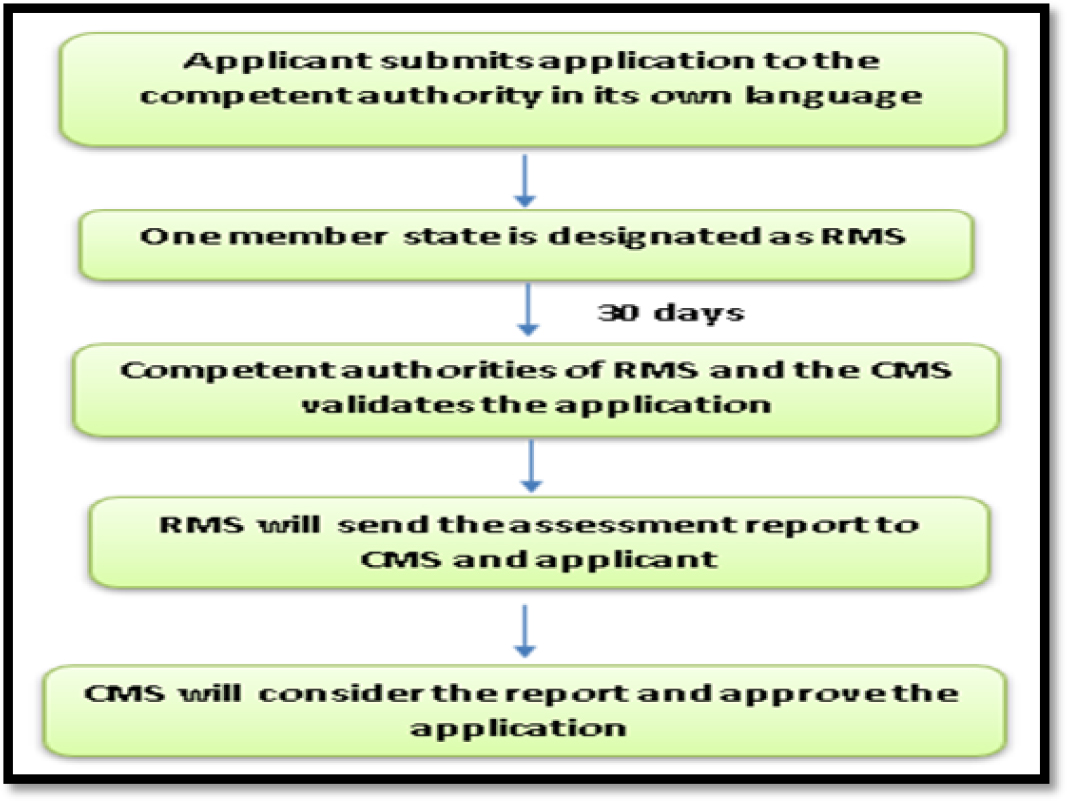
Figure 3:
National Procedure in EU for Generic drug approval.
Mutual Recognition Procedure
The process of mutual recognition procedure has been described in EU directive 93/39EEC. The ‘Reference Member State’ (RMS) in EU is the first Member State which assess the application. Other EU countries, were known as ‘Concerned Member States’ (CMS), where the applicant seeking the drug approval. Identical applications are submitted by applicant to both RMS and CMS. The submitted proposal will be validated by RMS within 90 days of initial submission. The assessment report made by RMS will be sent to CMS. CMS validates the application submitted by the applicant and RMS provides final assessment report within 90 days of RMS report submission. Once the application and assessment report has been validated by CMS, it approves the generic drugs. This is the only route available for generics, if the reference product authorized by national procedure.12
The Mutual Recognition Procedure for generic drug approval process in EU has been depicted in Figure 4.
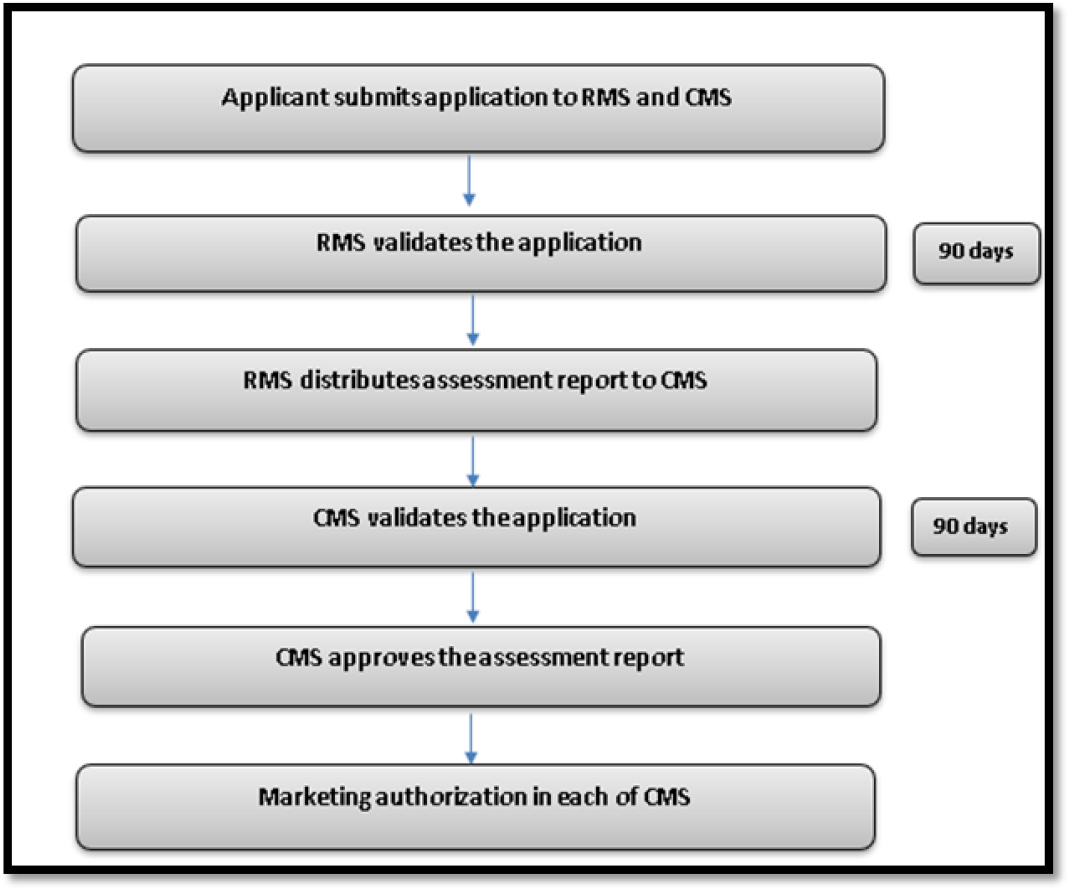
Figure 4:
Mutual recognition procedure in EU for Generic drug approval.
Decentralized Procedure
This regulatory approach is required for getting the marketing authorization in all the EU member states when no such marketing authorization has been granted previously for the generics in the EU. Initially the applicant submits the application to both RMS and CMS (s). Within 70 days of receipt of dossier, the RMS sends the preliminary assessment report in standard format to CMS (s). The CMS (s) are requested to provide comments on the proposed application and required to notify both applicant and RMS. On 105th day of initial submission of dossier, the RMS will send remarks to applicant and may pause the time, if necessary, until the applicant send required document to RMS on request. Once the required document has been received by RMS, it will prepare and compile a Draft Assessment Report and may approve the process of DP, if the CMS(s) and the RMS reach to an agreement.12,13
The Decentralized Procedure for generic drug approval process in EU is shown in Figure 5.
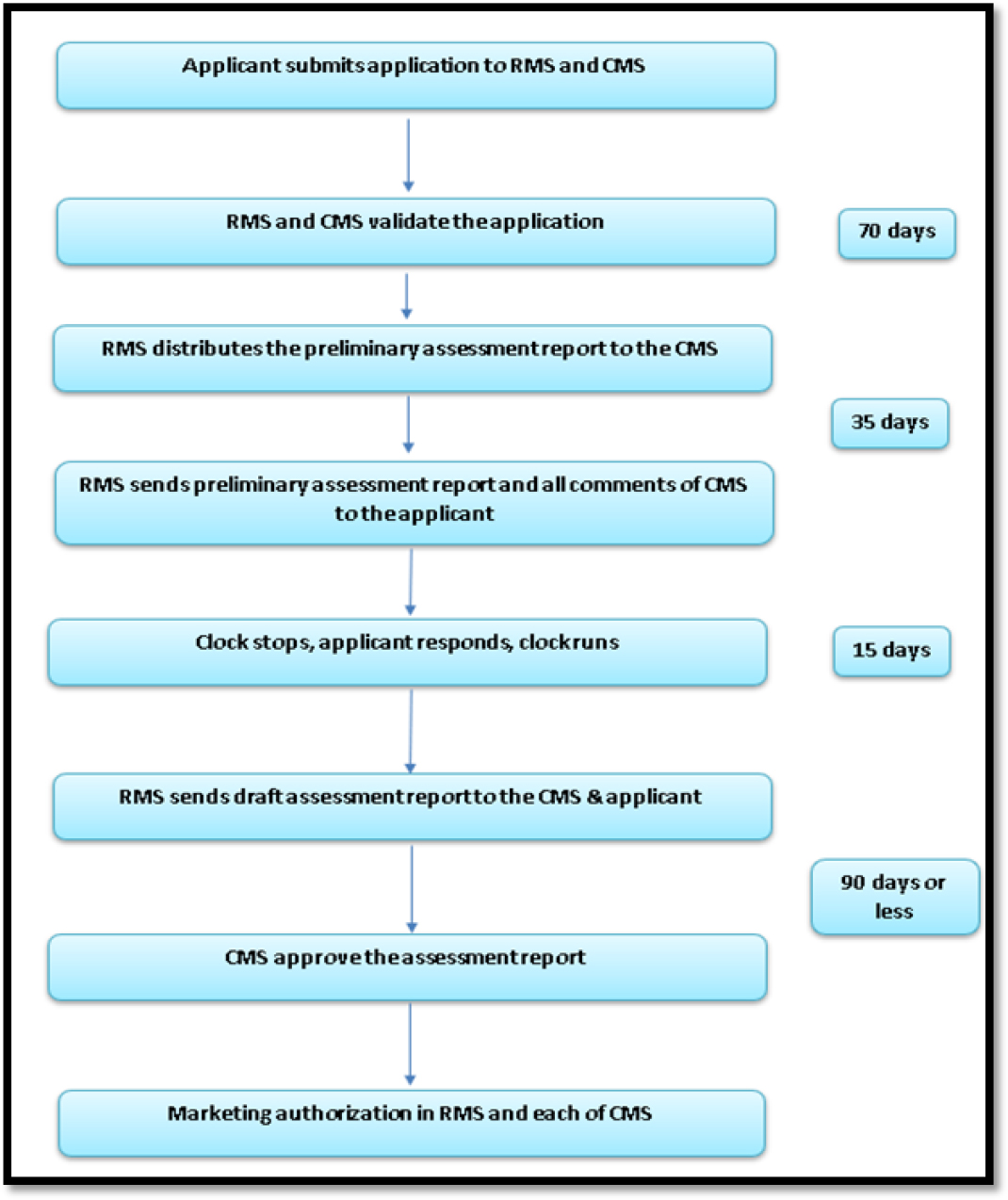
Figure 5:
Decentralized procedure in EU for Generic drug approval.
Generic drug approval process in UK
Since UK has been separated from EU through Brexit process, it follows the same generic drug approval process. Applicant submits the MAA to MHRA and the MAA document should be in English language only. MHRA evaluates the dossier and further asks for advice from its advisory committee. During the evaluation, if the submitted documents are incomplete, the authority will request the applicant to furnish further documents after 30 days of initial application submission. If all the submitted documents are satisfied by MHRA, it will produce an assessment report.14,15
The Generic drug approval process in UK has been depicted in Figure 6.
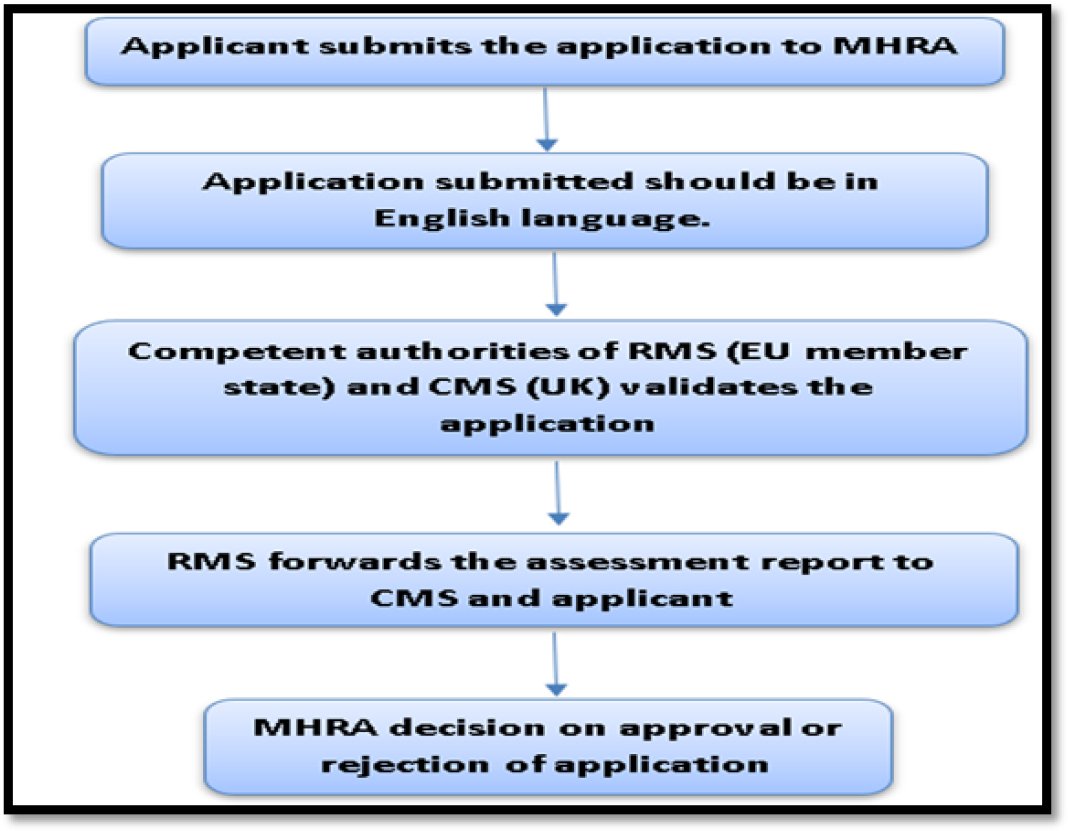
Figure 6:
Generic drug approval process in UK.
Generic drug approval process in Australia
The generic drug approval process in Australia involves 8 following steps.
Phase 1-Pre-submission: Pre-submission phase is the first step in the generic drug registration pathway. It applies to the generic drugs of category 1 and category 2. It involves the submission of Pre-Planning Form (PPF) along with application fees.
Phase 2-Submission of new generic drug application: This step involves the submission of new generic drug application which must be electronically submitted though electronic Business Service (eBS). Once the application is received by TGA, the applicant will be notified regarding the status of the submission whether the application has been on hold or under review or under acceptance. If the application kept on hold, the notification letter provides the reason for the same.
Phase 3-First Round Assessment: In this phase, the TGA reviewers will critically evaluate all the data and information present in the dossier
Phase 4-Consolidated section 31 request response: During the review if there are any quires regarding the dossier, TGA will send a request to the applicant to provide the response or answers for the questions raised and the applicant has to respond to the quires in ‘consolidated section 31 request’ form. ‘Consolidated section 31 request’ is a request sent by TGA to the applicant for providing information in case, if any questions are raised during the application evaluation, or if the submitted data is inadequate.
Phase 5-Second Round Assessment: Once the section ‘31 request form’ has been received from the applicant, the TGA evaluation team will verify the same and proceeds further for second round assessment of the revised dossier. Once the second-round assessment gets completed, TGA compiles the final report.
Phase 6-Expert advisory review: In addition to further review, the TGA may seek independent advice from its Advisory Committee on Medicines (ACM) to provide their opinion on the final report.
Phase 7-Decision: After the complete review of the dossier, the TGA expert team will declare whether approval for the proposed application can be granted or rejected.
Phase 8-Post-decision: Once the applicant is informed about the TGA decision, the post-decision phase begins. Administrative and regulatory activities are accomplished during this phase.16,17
The generic drug approval process in Australia is depicted in Figure 7.
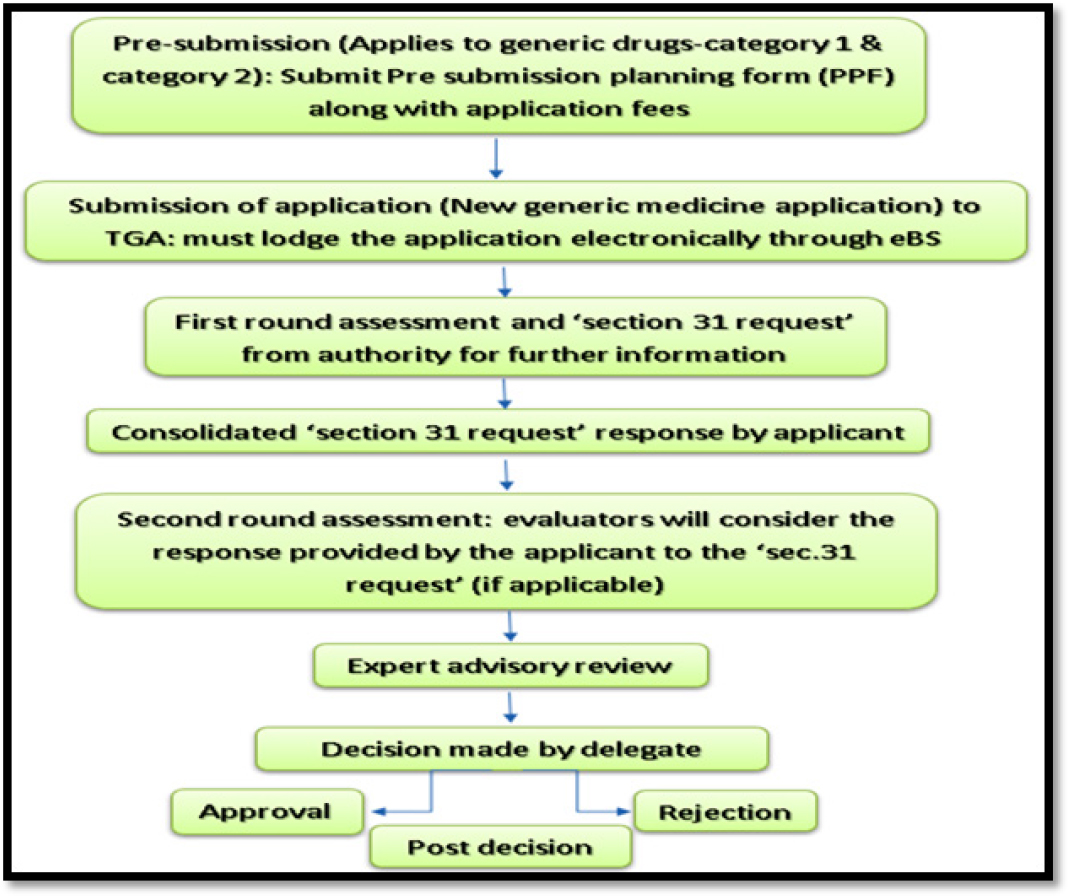
Figure 7:
Generic drug approval process in Australia.
Generic drug approval process in New Zealand
The applicant has to submit the Abbreviated New Medicine Application (ANMA) in English language to the MEDSAFE in the form of CTD. The submitted ANMA will be reviewed by advisory committees of MEDSAFE and provides advice to MEDSAFE regarding the application approval. In case, if the submitted information or documents found inadequate by the authorities, they will notify the applicant for providing further information which has to be furnished within 28 days. Once all the queries have been clarified by the applicant, the MEDSAFE seek further advice from advisory committee for the final decision. Application will be either accepted or rejected based on the decision of MEDSAFE review report.18,19
The generic drug approval process in New Zealand is shown in Figure 8.
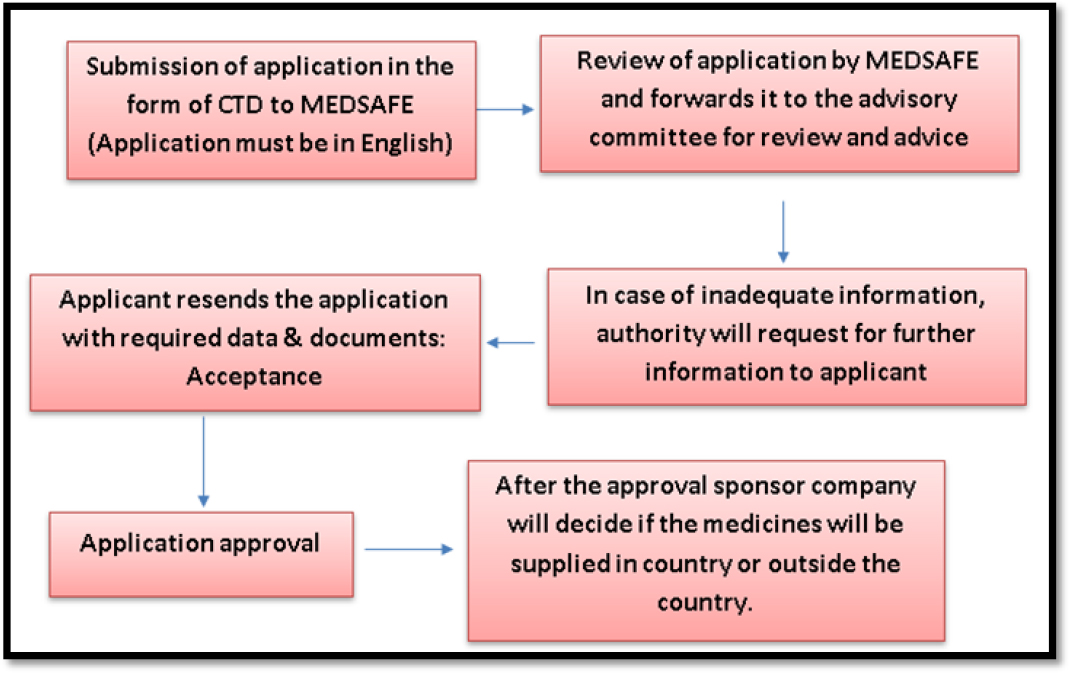
Figure 8:
Generic drug approval process in New Zealand.
CONCLUSION
Generic drugs that are identical copies of branded drugs, have been regulated and approved by drug regulatory authorities to make high-cost branded drugs available at cheaper cost to general public and are exported to other countries to increase the market revenue. Maintaining the quality and safety of generic drug is most challenging by generic drug companies and hence the regulatory authorities have put forth stringent regulations for generic drug approval. Generic drugs are approved by regulatory authorities only after 20 years of patent expiry. Each of these four countries has their own drug regulatory authority for regulating and approving the generic drugs. For generic drug approval, all four countries adopt the eCTD format, which facilitates a simple and speedy review process. In Europe, UK and Australia the generic drug application for marketing authorization is termed as MAA, whereas in New Zealand it is termed as Abbreviated New Medicine Application (ANMA). Generic drug application approval timeline in Europe and UK is same which takes nearly 1 year. The time period for Australian generic drug approval is 11 months, while it takes only 200 calendar days in New Zealand which shows that MEDSAFE evaluates the generic application quickly. Australia has set high price for generics while the New Zealand sets lowest price. All countries have regulations and guidelines for generic drugs, but still more stringent regulations are required to be framed for maintaining their quality and safety as that of branded drugs.
References
- [Jul 6 2022];Generic Drugs Overview [internet].

- Duerden MG, Hughes DA. Generic and therapeutic substitution in UK: are they a good thing. Br J Clin Pharmacol. 2010;70(3):335-41. [PubMed] | [CrossRef] | [Google Scholar]
- [Jul 6 2022];European Medicines agency. Generic medicine [internet]. [PubMed] | [CrossRef] | [Google Scholar]
- Birkett DJ. Generics equal or not. Aust Prescr. 2003;26(4):85-7. [CrossRef] | [Google Scholar]
- Lockhart MM, Babar ZU, Carswell C, Garg S. New Zealand’s drug development industry. Int J Environ Res Public Health. 2013;10(9):4339-51. [PubMed] | [CrossRef] | [Google Scholar]
- Mumeena S. Comparison of regulatory requirements for generic drugs dossier submission in United States, Europe and Canada. Int J Pharm Chem Res.. 2017;3(2):370-96. [PubMed] | [CrossRef] | [Google Scholar]
- [Jun 16 2022];Apply for a license to market a medicine in the UK [internet]. [PubMed] | [CrossRef] | [Google Scholar]
- [Jun 16 2022];Strategies for generic/biosimilar market entry in Australia [internet]. [PubMed] | [CrossRef] | [Google Scholar]
- Kumari BS, Hanuja GS, Nagabhushanam MV, Reddy DN. Current regulatory requirements for registration of medicines compilation and submission of dossier in Australian Therapeutic Goods Administration. Int J Adv Sci Res.. 2017;6(6):144-56. [PubMed] | [CrossRef] | [Google Scholar]
- [Jun 16 2022];New Zealand looks to revise its abbreviated process policy [internet]. [PubMed] | [CrossRef] | [Google Scholar]
- Gauld NJ, Emmerton L, Kelly F, Buetow S.. Widening consumer access to medicines: a comparison to non-prescription medicine switches in Australia and New Zealand. PLOS ONE. 2015;10(3):1-22. [PubMed] | [CrossRef] | [Google Scholar]
- Jawahar N, Datchayani B. Comparison of generic drug application and their approval process in US, Europe and Japan. J Pharm Sci Res.. 2018;10(3):523-27. [PubMed] | [CrossRef] | [Google Scholar]
- Abed I. The approval process of medicines in Europe. J Am Writ Ass.. 2014;23(2):117-21. [CrossRef] | [Google Scholar]
- Sandeep DS, Koppal S, Charyulu RN, Nayak P.. Drug registration and approval process in United Kingdom. Pharma Times. 2018;50(8):20-2. [CrossRef] | [Google Scholar]
- [Apr 30 2022];Apply for a license to market a medicine in the UK [internet]. [CrossRef] | [Google Scholar]
- [May 25 2022];TGA Prescription medicines registration process [internet]. [CrossRef] | [Google Scholar]
- Hasan SS, Kow CS, Dawoud D, Mohamed O, Baines D, Babar ZU, et al. Pharmaceutical policy reforms to regulate drug prices in the Asia Pacific Region: the Case of Australia, China, India, Malaysia, New Zealand and South Korea. Value Health Reg Issues. 2019;18:18-23. [PubMed] | [CrossRef] | [Google Scholar]
- [Apr 25 2022];Guideline on the regulation of therapeutic products in New Zealand [internet]. [PubMed] | [CrossRef] | [Google Scholar]
- Gauld NJ, Emmerton L, Kelly F, Buetow S.. Widening consumer access to medicines: a comparison to nonprescription medicine switches in Australia and New Zealand. PLOS ONE. 2015;10(3):1-22. [PubMed] | [CrossRef] | [Google Scholar]